Hi
I’m working on simulation of evaporation and I want to separate evaporated atoms from liquid ones. To do this I want to use Oxford method. According to the Oxford method, the radial distribution function is adopted to distinguish the liquid and vapor molecules. A liquid–vapor equilibrium system is used to calculate the radial distribution function. Molecule i is recognized to be in the liquid phase, if it has at least N (the number integrating of the radial distribution function from zero to the first valley point) neighbor molecule j.
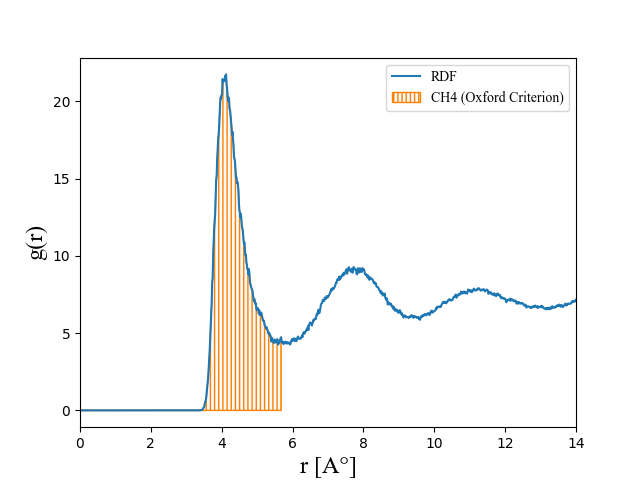
To do this I let my system to reach a liquid–vapor equilibrium system and then I calculate the RDF. But the result is not rational(as you can see below).
With respect to this RDF a molecule would count as liquid if surrounded at least by 22 molecules only with 6 angstrom distance!
I want to know if this problem comes from the parallel computation. I mean if RDF must calculate in serial computing?
What problem? The RDF looks like a very good match.
No, the RDF calculation can be well parallelized. It uses the neighbor lists that LAMMPS also uses for computing forces from pair styles.
Where do you get the number 22 from? The absolute value of the g(r) graph has no direct meaning since it is not normalized. You have to plot the next column of the output, which has the number integral versus distance (by simply counting neighbors…).
With respect to this RDF a molecule would count as liquid if surrounded at least by 22 molecules only with 6 angstrom distance!
Is it possible?
The RDF output has three columns. You mean I have to integral over the third column values instead of second?
Please see: compute rdf command — LAMMPS documentation