Thanks for your responses Dr. Kohlmeyer and Dr. Dylan.
Lammps version is- 16 March 2018.
I am attaching the input below. I would request you to take a quick look. There are 5 metals in my system and I am trying to calculate the RDF between all like pairs and unlike pairs.
units metal
dimension 3
boundary p p p
atom_style atomic
variable latparam equal 3.21
----------------------- ATOM DEFINITION ----------------------------
lattice bcc {latparam}
region whole block 0 31.5 0 31.5 0 31.5
create_box 5 whole
lattice bcc {latparam} orient x 1 0 0 orient y 0 1 0 orient z 0 0 1
create_atoms 1 region whole
variable fa equal 12080
variable fb equal 9368
variable fc equal 3098
variable fd equal 1547
variable ft equal 62559
set type 1 type/fraction 2 (v_fa/v_ft) 1734536
set type 2 type/fraction 3 (v_fb/(12080)) 1734535
set type 3 type/fraction 4 (v_fc/(9368)) 1734534
set type 4 type/fraction 5 (v_fd/(3098)) 1734533
group Mo type 1
group W type 2
group Ta type 3
group Ti type 4
group Zr type 5
pair_style eam/alloy
pair_coeff * * MoTaWTiZr.set Mo Ta W Ti Zr
min_style cg
min_modify dmax 0.1
thermo 10000
thermo_style multi
minimize 1e-15 1e-15 1000000 1000000
compute csym all centro/atom bcc
compute peratom all pe/atom
reset_timestep 0
timestep 0.001
velocity all create 4000 123456 dist gaussian
fix 123 all npt temp 4000 4000 1 iso 0 0 1 drag 1
thermo 1000
thermo_style custom step temp press ke pe etotal vol density
run 90000
compute myRDFMo_Mo all rdf 200 1 1
fix 101 Mo ave/time 100 1 100 c_myRDFMo_Mo[*] file tmp1_Mo.rdf mode vector
compute myRDFW_W all rdf 200 2 2
fix 102 W ave/time 100 1 100 c_myRDFW_W[*] file tmp1_W.rdf mode vector
compute myRDFTa_Ta all rdf 200 3 3
fix 103 Ta ave/time 100 1 100 c_myRDFTa_Ta[*] file tmp1_Ta.rdf mode vector
compute myRDFTi_Ti all rdf 200 4 4
fix 104 Ti ave/time 100 1 100 c_myRDFTi_Ti[*] file tmp1_Ti.rdf mode vector
compute myRDFZr_Zr all rdf 200 5 5
fix 105 Zr ave/time 100 1 100 c_myRDFZr_Zr[*] file tmp1_Zr.rdf mode vector
compute myRDFMo_W all rdf 200 1 2
fix 106 all ave/time 100 1 100 c_myRDFMo_W[*] file tmp1_MoW.rdf mode vector
compute myRDFMo_Ta all rdf 200 1 3
fix 107 all ave/time 100 1 100 c_myRDFMo_Ta[*] file tmp1_MoTa.rdf mode vector
compute myRDFMo_Ti all rdf 200 1 4
fix 108 all ave/time 100 1 100 c_myRDFMo_Ti[*] file tmp1_MoTi.rdf mode vector
compute myRDFMo_Zr all rdf 200 1 5
fix 109 all ave/time 100 1 100 c_myRDFMo_Zr[*] file tmp1_MoZr.rdf mode vector
compute myRDFW_Ta all rdf 200 2 3
fix 110 all ave/time 100 1 100 c_myRDFW_Ta[*] file tmp1_WTa.rdf mode vector
compute myRDFW_Ti all rdf 200 2 4
fix 111 all ave/time 100 1 100 c_myRDFW_Ti[*] file tmp1_WTi.rdf mode vector
compute myRDFW_Zr all rdf 200 2 5
fix 112 all ave/time 100 1 100 c_myRDFW_Zr[*] file tmp1_WZr.rdf mode vector
compute myRDFTa_Ti all rdf 200 3 4
fix 113 all ave/time 100 1 100 c_myRDFTa_Ti[*] file tmp1_TaTi.rdf mode vector
compute myRDFTa_Zr all rdf 200 3 5
fix 114 all ave/time 100 1 100 c_myRDFTa_Zr[*] file tmp1_TaZr.rdf mode vector
compute myRDFTi_Zr all rdf 200 4 5
fix 115 all ave/time 100 1 100 c_myRDFTi_Zr[*] file tmp1_TiZr.rdf mode vector
compute myRDFall_1 all rdf 200
fix 116 all ave/time 100 1 100 c_myRDFall_1[*] file tmp1_all.rdf mode vector
The output from the last line (rdf for all atoms) in the above code gives a radius ‘r’ ranging from 0 to >80 angstroms -
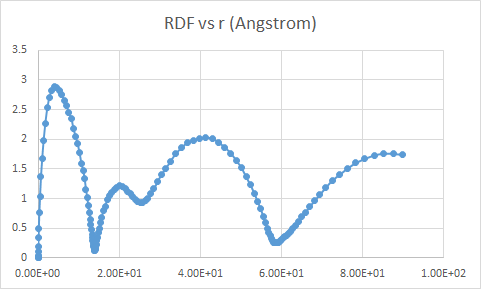
The output from all other RDF computations is similar to this figure below, with x axis range from 0 to ~2 angstroms.
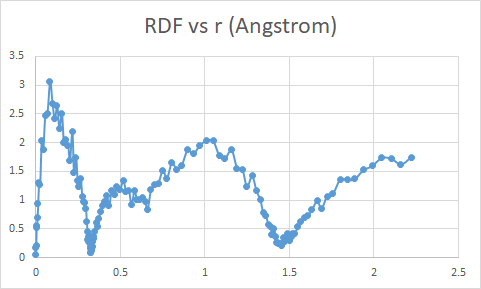
My question is - is there a way to make the radius ‘r’ range same for both RDF of all atoms and RDF of specific groupwise atoms ?