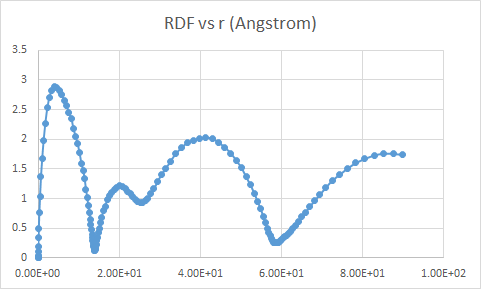
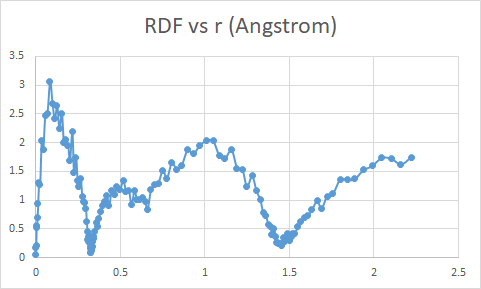
you are interpreting the data in the .rdf files incorrectly.
the first column is the bin number, the second column is radius, the third column is the RDF and the fourth column is the integrated RDF (which is the number of neighboring atoms up to the value of r.
you seem to be interpreting the integrated RDF as radius.
the RDF data is in all cases computed from 0 to 7.15537, the force cutoff of your EAM potential.
if you look at your plots, they look completely unreasonable. there should be excluded volume of the atoms for at least about 2 angstrom and thus the RDF zero or near zero.
please also note, that it is not necessary, to have this many compute rdf commands, all the different computations of RDFs of type pairs can be done in a single compute RDF statement.
please have a look at the documentation again.
this is entirely a user issue. LAMMPS is working correctly and as documented.
axel.