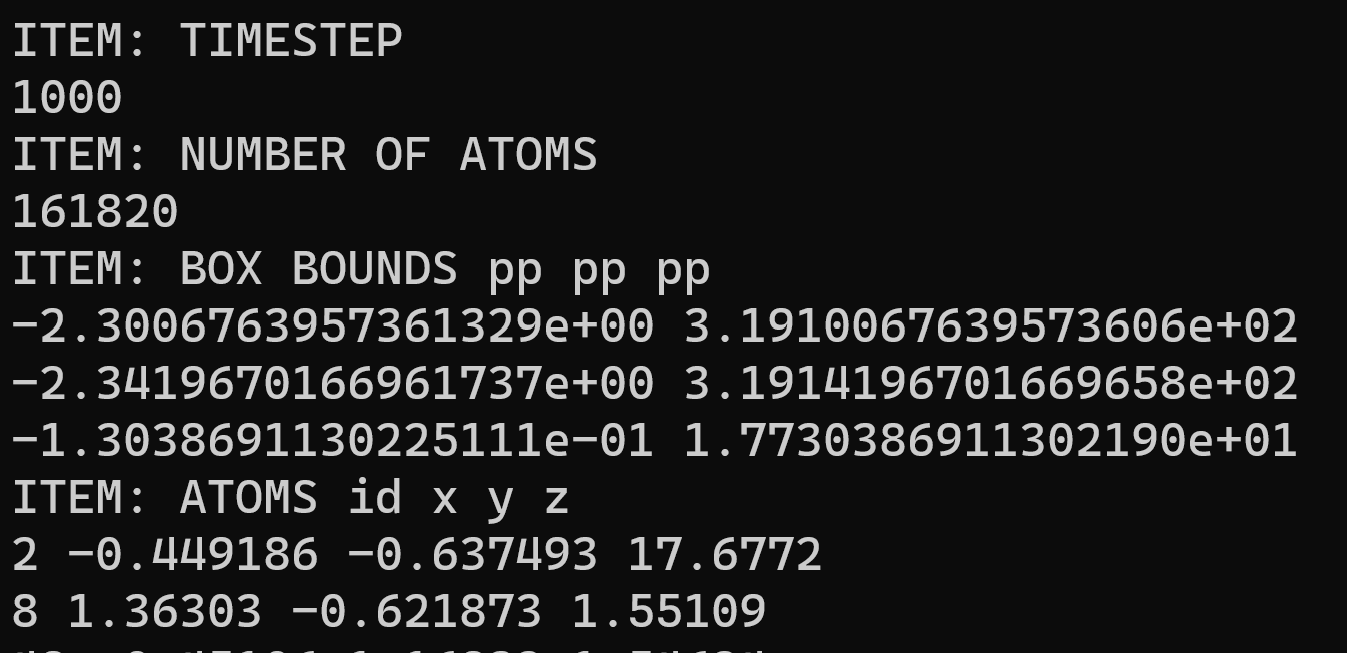
I tried using the read_dump command to read in the atom positions, but it’s not working:
ERROR on proc 0: Read_dump field not found in dump file (src/lammps/src/read_dump.cpp:533)
Last command: read_dump avg-1000.dump 1000 x y z add yes
What is that I’m doing wrong with this command? Thank you!
When reporting errors, please always report which specific LAMMPS version you are using and what platform you are running on, as this may be crucial information to determine the cause of the error.
If you are using the “add yes” flag, you tell LAMMPS to create new atoms, but that is not possible without setting an atom type. But your dump file only contains the atom ID and the positions, so read_dump has to error out and report that a field is missing (i.e. the “type” field).
This is a very inexact (and thus unscientific) description. What is “the most recent” depends on 1) when you last checked/updated, 2) whether you picked a download version or cloned a git repo branch, 3) if the former whether you downloaded a stable or a feature release version, 4) if the latter, which of 4 available branches you have checked out, 5) whether you use a pre-packaged or auto-match built version and when this was last updated and so on. I have seen this cover a range of more than 3 years at times.
The most accurate (and thus scientific) description is to quote the first couple of lines from the lmp -h output.