Dear all,
Actually to build my model I have to read many data file(say, 50).So instead of using many add/append/offset,if I keep all those data file in a folder and give the folder name or any command using folder name in my LAMMPS input script then it willbe relatively easy. I have read read data command.So is there any process to do that?
Thanks…
Your question is not very clear, but it seems that you don’t want to have an input file with a long list of read_data statements. Suppose your data files are named in1.data, in2.data, etc, you can use a loop like this:
variable i loop 50
label input_data
read_data my_folder/in${i}.data append merge # modify as necessary
next i
jump SELF input_data
I would recommend doing this in two steps.
Firstly, use the Linux shell to make copies of your data files with formulaic, sequential numbered names. For example:
ls -v | grep '.data' | cat -n | while read n f; do cp -n "$f" "DATAFILE$n"; done
this will copy the first .data file into DATAFILE1, the second .data file into DATAFILE2, and so on. (Script modified from StackOverflow: rename files with sequential numbers.)
Then use LAMMPS’s loop variables and next command to loop through the data files.
1 Like
Thank you sir.
Yes sir, I don’t want to have an input file with a long list of read_data statements.
As you told I have used that in the following code with some modification.But it’s showing illegal read_data command.Actually this process I had followed earlier,then it was right,except that loop.Now,how can I deal with extra atom/bond types after using this loop?Because I have to make space for them earlier.
Thanks.
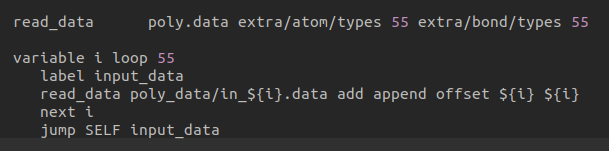
Thank you very much.
Rather than discussing technical solutions for your specific question, can you first please explain the scientific reason for why you think you have to follow this approach and how you create all those many data files.
I suspect that there may be alternatives that could be simpler to follow.
Sir, suppose I want to create 100 polymers consisting of different no of monomers.So for those 100 polymers I have 100 separate data file and I want to read those data file in LAMMPS input script in a simple way. Using python we can create data for those 100 polymers in only one data file, but I want any other simple ways.
If there are any alternatives then please help me.
So how do you obtain or create those 100 data files?
Also, what kind of polymers are you talking about? Simple bead-spring models or specific all-atom models or coarse grained models etc.
Technically, you can read multiple data files with “read_data xxx add append”, but there is no wild card processing for filenames, so you would have to generate the sequence of LAMMPS input commands with an external tool (e.g. a python script) so you can then use “include” to have LAMMPS process those commands.
However, I fail to see how that would be simpler than just creating the whole system with a custom tool in the first place. For example, how would you make sure that there are no or not many overlaps? And how would you then resolve the overlap situation? How would you manage equilibraton considering that polymers are usually quite entangled and achieving that from individual monomers can be quite complex?
So, you need to elaborate in much more detail what you are aiming to do and specifically how using read_data on hundreds of data files is going to make things simpler. It still sounds a lot like you have a case of “premature optimization”, i.e. worry about something that is not having much impact in total on your work.
To complete on what @akohlmey is saying, rather than data files, consider looking at molecule files.
Using create_atoms command you can insert several molecules from the same file with random orientation and avoid overlap (to some extend) at given or random positions. It still require careful checking of each insertion in the log files but they might be easier to make from scripting (no box definition). It might also help in reducing the bond and atom types by consistently using the same in the different molecule files.
However, Axel’s remark on the making of bulk polymer systems are still valid so this will replace thinking on your systems creation in no way.
1 Like