Hi.
I tried validating the ffield.reax.rdx using the 21st July 2010
version of LAMMPS compiled with the FORTRAN reax library (the C
version was probably not available then). The ffield.reax.rdx from the
README.reax are the parameters used in [Zhang, L., van Duin, A.C.T.,
Zybin, S. and Goddard, W.A. (2009) Thermal Decomposition of Hydrazines
from Reactive Dynamics Using the ReaxFF Reactive Force Field, Journal
of Physical Chemistry A 113, 10770-10778]. In the "examples/reax/RDX"
directory of the April-5^th version these are same as the parameters
used by A. Strachan et al. [Phys. Rev. Lett. 2003, 91, 098301].
I tried to obtain the unit cell sizes of crystal RDX by carrying out a
"fix NPT" run with the temperature increasing from 50 K to 300 K and
pressure maintained at 0 atm in the first 100 pico-secs. Then for the
next 200 pico-secs the temperature was maintained constant at 300
pico-secs with the pressure kept at 0 atm. I used a 2x2x2 unit cells
of RDx (with 1344 atoms and a time step of 0.1 femto-sec - see
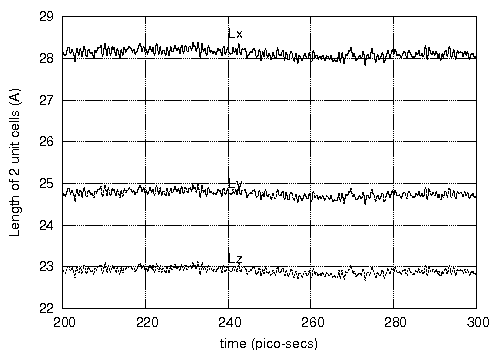
attached input file). Averaging over the sample sizes along the X
(Lx), Y (Ly) and Z (Lz) I get Lx = 14.04, Ly = 12.36 and Lz = 11.43
Angstroms (see attached figure LxLyLz.eps - note that these are for a
2x2x2 unit cell sample so I divide the average value of the
equilibrated fluctuations by 2). These however differ from the values
reported by A. Strachan et al. (Lx = 13.78, Ly = 12.03 and Lz = 10.96
Angstroms) [Phys. Rev. Lett. 2003, 91, 098301].
The above difference are not "LARGE" considering that the MD method
used to obtain the unit cell sizes are different, but I also note that
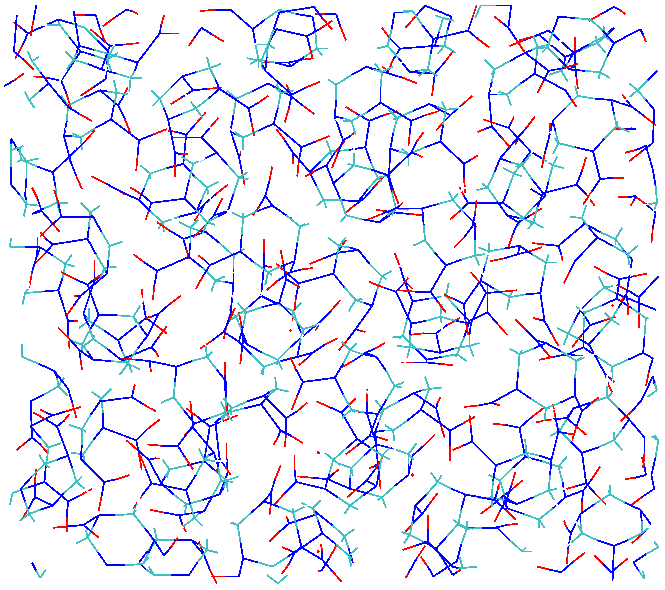
the structure of crystal RDX seems to have changed (this happens
around 150 - 180 K during the ramp up of temperature carried out in
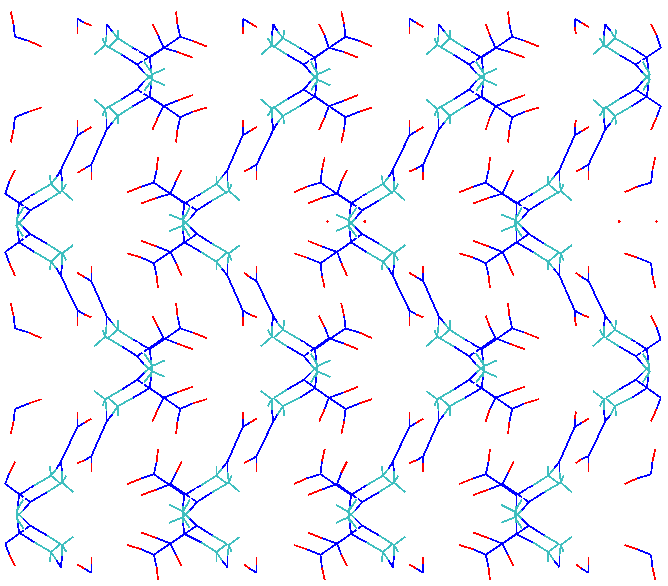
the first 100 pico-seconds. I am attaching a vmd rendering of the
2x2x2 RDX crystal before (2x2x2xRDXInitial.eps) and after
(2x2x2RFFNFinal.eps) the simulation.
Therefore can someone tell me if there is some mistake in my input
file? Is the structural change expected? I have not simulated
molecular crystals earlier and are there any special considerations I
should make when doing so?
Please excuse the poor quality of images attached. I had to make them
low resolution to squeeze into the size limits of mails not needing
moderator approval in this mailing list.
Thanking you.
With Best Regards
Manoj
in.reax.rdx (910 Bytes)