I am trying to simulate Orcinol crystal. To calculate the free energy we
followed Einstein molecule method, in which each atom is attached to its
lattice site. we used fix spring/self for this purpose. If i want to
restart my simulation using restart files, The atom positions were
changed, but ineed to keep my reference at lattice positions only for the
new simulation.
How can i implement it in lammps?
RAVI KUMAR REDDY A
Research Scholar
IISc Bangalore.
I am trying to simulate Orcinol crystal. To calculate the free energy we
followed Einstein molecule method, in which each atom is attached to its
lattice site. we used fix spring/self for this purpose. If i want to
restart my simulation using restart files, The atom positions were
changed, but ineed to keep my reference at lattice positions only for the
new simulation.
How can i implement it in lammps?
fix spring/self supports restart files and stores the original
positions in them. when restarting, you should see a message, that fix
spring/self is restoring its internal data from the restart.
if you have reason to believe, that this doesn't work correctly,
please provide a minimal input example demonstrating the failure.
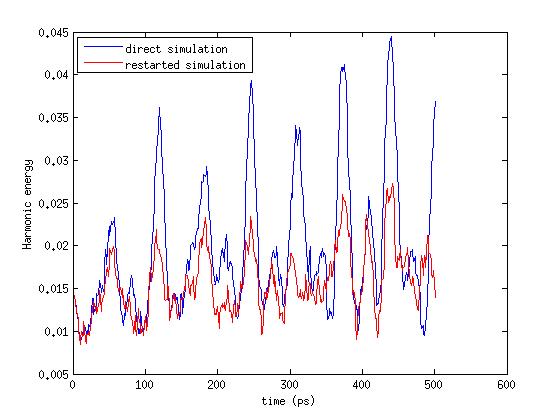
I read same about spring/self that which will keep original coordinates in
restart files but i observed the different dynamics in both cases.
I ran 1000000 time steps of simulation in which i saved my restart files
at every 500000 time steps. In second case i used restart file at 500000
step and ran it. The log files (harmonic spring energies) were different
in both cases. I am attaching my input script and log files here.
I have plotted harmonic energy from spring/self in both cases, that file
is also attached here
please have a look at it and give your comments
RAVI KUMAR REDDY A
Research Scholar
IISc Bangalore
I read same about spring/self that which will keep original coordinates in
restart files but i observed the different dynamics in both cases.
this has nothing to do with fix spring/self. as you can see, the
energy is identical right after the restart. that would not be the
case, if fix spring/self would use new origin coordinates.
I ran 1000000 time steps of simulation in which i saved my restart files
at every 500000 time steps. In second case i used restart file at 500000
step and ran it. The log files (harmonic spring energies) were different
in both cases. I am attaching my input script and log files here.
I have plotted harmonic energy from spring/self in both cases, that file
is also attached here
this divergence is expected. writing/reading a restart enforces a
neighbor list rebuild, which can lead to a reordering of atoms in the
neighbor list due to migration to different subdomains or across
periodic boundaries. since floating point math is not associative, the
order in which atom forces matters and thus you'll get small
differences due to truncation and roundoff and trajectories will
eventually diverge exponentially and be fully decorrelated.
this is a well known property of MD simulations with floating point
math and has been discussed on this very mailing list regularly. it is
also mentioned in the manual and should be addressed in good text
books on MD.