Hello Everyone,
I had a question about the "replicate" command in LAMMPS. If I carry 3-D MD
simulations on a graphene-like single-layered structure like MoS2
(Molybdenum Suilphide), can I create a single molecule of that structure
using a software,
then create its data file in LAMMPS readable format and
then use replicate command to create similar such hexagonal molecules (so as
to form a sheet of atoms like Graphene) and then carry out the simulation?
Will this give the right results?
Although I'm not sure this reply will help you, I never miss an
opportunity to shamelessly promote "moltemplate"!
In general, if you have an existing LAMMPS data file and input script,
and you want to extract a molecule from it, replicate it, and move it
to a new position see this post:
(More detailed instructions for this example are located in the
tools/moltemplate/examples/convert_LAMMPS_to_LT_examples/cnad-cnt/
directory distributed with lammps.)
If you have a LAMMPS data file and you want to duplicate everything in
that file, (or merge it with another data file), then see this post:
These examples use the "ltemplify.py" utility to convert an existing
LAMMPS data-file+input-script into a single .LT file. LT files are
intended to be easily readable by humans, but an LT file created by
ltemplify.py will be difficult to read (although it contains the same
information). If you have a little more time, you can try to build
the .LT from scratch.
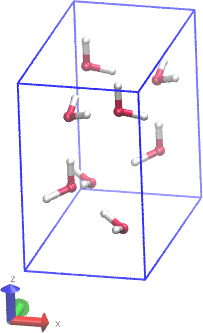
You can define a unit cell for a crystal, and duplicate it to build a
larger crystal. The atoms in each unit cell can contain bonds,
angles, etc.
See the example this directory:
tools/moltemplate/examples/all_atom_examples/ice_crystal
(also distributed with lammps)
However currently, the bonds can not connect atoms in different unit
cells together. (Topotools can get around this particular
restriction, but even with topotools, bonds currently can not cross
the periodic boundary conditions for the entire simulation box, as
Axel said.)
in fact, you can do the *entire* process in LAMMPS (or entirely with
external tools) for as long as you don't need to add explicit bonds
(and angles, dihedrals, impropers). this is where it gets tricky,
since bonding across periodic boundaries doesn't agree with the
replicate command and practically all external packages.
There may be ways to do many of these things using only LAMMPS input
script commands, but moltemplate and topotools give you some options
which LAMMPS input script commands don't. (Such as rotation and
customization. Moltemplate objects (LT files) are very portable and
make it easy to combine different molecules and different force-fields
together in the same simulation.)
My boundaries will be periodic where there are no explicit bonds between
neighbors. The atoms will be modeled with some potential as there are no
permanent bonds.
so there is no problem. define a suitable lattice, create a region for
your box, a region for your sheet and fill it with atoms. done.
Yep.
Either moltemplate or topotools or lammps input script commands should
work fine. My apologies for not providing instructions for topotools
or lammps. (I am more familiar with moltemplate.)
I read the manual but was a bit confused about this and so will be very
helpful if someone can please confirm. Thank you so much.
In addition to moltemplate and topotools, take a look at:
Cheers
Andrew