Are you certain that you need to have bonds in your crystal?
(See "Comments" at the end of this email.)
It is surprisingly difficult to do generate these kinds of bonds in moltemplate
(even if you write a for-loop to generate them. See comment #2 below...)
Moltemplate is currently does not yet have the ability to efficiently
check whether two atoms are close together, and add connecting bonds.
If you are certain that you need to simulate crystals filled with bond
constraints, then I recommend that you use use topotools to assign
bonds between nearby atoms. Then, if you still wish to use
moltemplate, you can convert the crystal created by topotools
(including atoms, bonds, angles, etc...) into a moltemplate object
format using the "ltemplify.py" utility.
------------------------ Explanation ----------------------
(This was written in a hurry.
Please correct me if you find any mistakes.)
If you already have created the crystal in moltemplate, but it
lacks bonds, then you can use moltemplate to convert it into a DATA
file.
moltemplate.sh system.lt #(this creates "system.data")
Then use topotools to read the DATA file created by moltemplate
(typically called "system.data" using these commands (typically run
from inside the "Tk Console" in VMD):
topo readlammpsdata system.data full
(Note: you don't need to use moltemplate to generate the crystal.
Topotools has its own commands. See link below.) After you have
created or loaded the crystal into topotools, then use this command to
generate bonds between nearby atoms:
set sel [atomselect top all]
$sel set radius 2.1
mol bondsrecalc top
I have not tried this. You may have to to play with the radius
parameter to get this to work. For more details, see:
https://sites.google.com/site/akohlmey/software/topotools/topotools-tutorial---part-1
https://en.wikipedia.org/wiki/Atomic_radii_of_the_elements_(data_page)
Next, write out a new DATA file using this command
topo writelammpsdata crystal.data angle
Finally, use ltemplify.py to create a new .lt file.
echo "atom_style full" > crystal.in
Then run ltemplify.py to convert crystal.data into a moltemplate file:
ltemplify.py -name Crystal crystal.in crystal.data > crystal.lt
Finally, create a new .LT file, and add these commands to that
file (in addition to other molecules you wish to use, like water):
import crystal.lt
crystal = new Crystal
You can add other molecules to this file, for example, water:
import "spce.lt"
wat = new SPCE [10].move(0.00, 0.00, 3.45)
[10].move(0.00, 3.45, 0.01)
[10].move(3.45, 0.01, 0.01)
Finally, run moltemplate.sh on this new .LT file.
ltemplify.py is explained in appendix B of the moltemplate manual:
http://www.moltemplate.org/doc/
An example of ltemplify.py usage here:
http://www.moltemplate.org/examples/nanotube+water/chiral_nanotubes.html
------- COMMENTS: --------
I have never attempted a simulation of minerals
1) If you simulating pyrophyllite crystals in water at low
temperatures, then it is really the water you are simulating. You
probably don't need to use an accurate force-field if the atoms in the
pyrophyllite, if they do not move very much. In that case, simply
omit all of the crystal atoms from the group of atoms being integrated
in "fix nvt" or "fix npt". Alternately, use something like "fix
restrain" or "fix rigid" to constrain their motion.
http://lammps.sandia.gov/doc/fix_restrain.html
http://lammps.sandia.gov/doc/fix_rigid.html
(If your crystal is as large as your simulation box, then you will
have to be careful when running simulations under NPT conditions.
http://www.moltemplate.org/examples/nanotube+water/run.in.nvt
Be warned that the advice in that link is no longer up-to-date, and
may no longer work.)
2) If you are certain that you must bonds to connect atoms between
unit cells, you could try using a for-loop to create bonds between
atoms in neighboring unit cells. Prompted by your email, I attempted
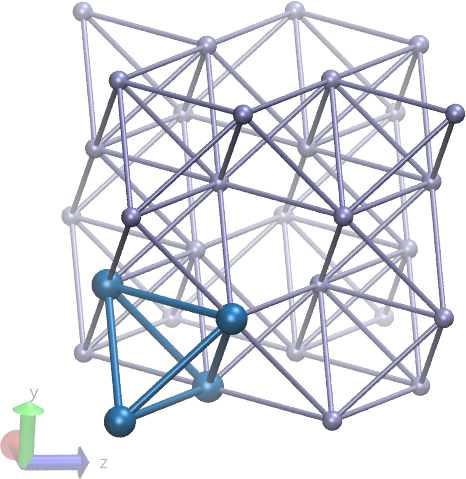
to make a bonded aluminum crystal. As a cautionary tale, I have
attached a picture of what this looks like. (...along with the python
script which generated them. Atoms in the first unit cell are shown
in dark blue. Apparently not all bonds are between nearest-neighbor
lattice sites. Notice missing bonds between atoms in unit cells
accross the central diagonal. It's certainly possible to change the
python script to add these diagonal bonds, but is it worth the
effort?)
In summary, I recommend you use topotools to generate the crystal, and
using ltemplify.py to convert the crystal back into moltemplate
format.
Even if you get around this issue, keep in mind that you will not be
able to simulate crystal dislocations and defects which may be
important at higher temperatures and/or pressures. Bonds behave like
harmonic constraints which prevent those kinds of atom movements from
occurring. However, LAMMPS offers several manybody pair_styles which
might be useful under these conditions. (To be clear, if you are
using these pair_styles, you should not be connecting these atoms
together with bonds.)
I hope this helps
Andrew
al_cell.lt (1.69 KB)
al_crystal_bonds.lt (5.06 KB)
al_crystal_bonds.py (5.84 KB)
system.lt (947 Bytes)