Dear lammps-users,
I am trying to study the minimum energy path using NEB algorithm between 2 stable states of Alanine dipeptide in lammps-7th Aug 2019 version.
From research articles available online, I got the values of phi, psi torsional angles in 2 different stable conformations of alanine dipeptide. I generated minimized alanine dipeptide structures with phi, psi values close to these 2 available conformations. Then I ran NEB between these 2 stable conformations.
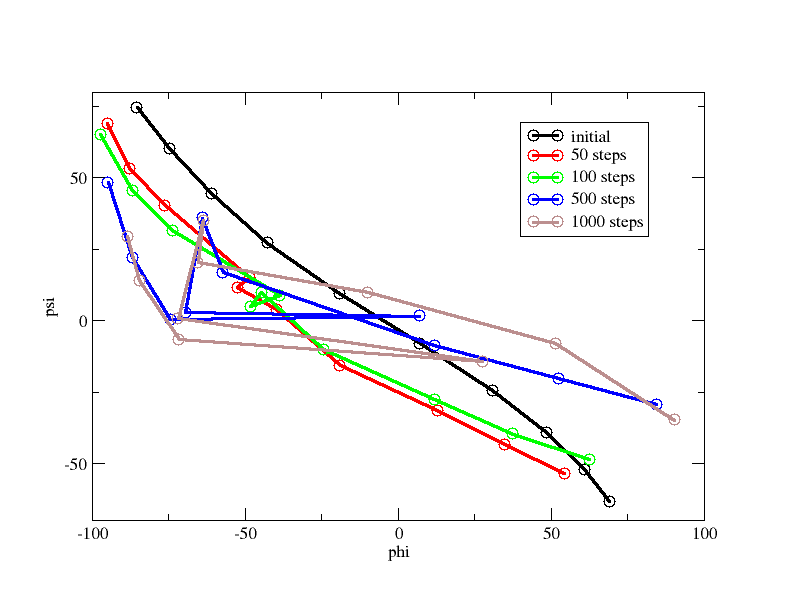
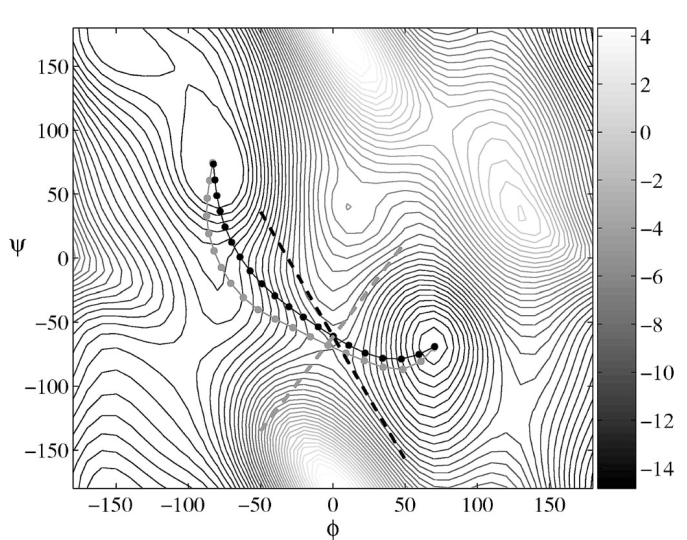
I see that as NEB runs go on, the phi, psi values for my first and last replicas change a lot from their starting values, which they shouldn’t, as they are already close to the minima. I also observe that the minimum energy curve I obtain (by plotting phi, psi values of replicas, attached image adp_neb.png, after 50, 100, 500 and 1000 neb steps) is very different from what is available in research papers (attached image mep.png, taken from String method in collective variables: Minimum free energy paths and isocommittor surfaces; Luca Maragliano, Alexander Fischer, and Eric Vanden-Eijnden, 2006). I have attached my input file in this mail as well.
It would be great if anyone could give me any insights into why this is happening.
Thanks in advance,
Madhur Aggarwal
CCNSB, IIIT Hyderabad
neb.inp (800 Bytes)