Hello everyone, I am a relatively amateur user of Lamps software, I am producing copper/graphene composite with Lamps software, the problem is that during the process, the set of copper nanoparticles (4 upper particles together and four lower particles) (together) constantly move on the graphene sheet and the more I increase the relaxation time, it has no effect, my code is as follows =
dimension 3
boundary p p p
units metal
atom_style atomic
#NanoParticles
read_data data1.txt
lattice fcc 3.615
region box1 sphere 23.85599 25.339 -29.7 25 units box
create_atoms 2 region box1
region box2 sphere 73.85599 25.339 -29.7 25 units box
create_atoms 2 region box2
region box3 sphere 123.85599 25.339 -29.7 25 units box
create_atoms 2 region box3
region box4 sphere 173.85599 25.339 -29.7 25 units box
create_atoms 2 region box4
region box5 sphere 23.85599 75.339 -29.7 25 units box
create_atoms 2 region box5
region box6 sphere 73.85599 75.339 -29.7 25 units box
create_atoms 2 region box6
region box7 sphere 123.85599 75.339 -29.7 25 units box
create_atoms 2 region box7
region box8 sphere 173.85599 75.339 -29.7 25 units box
create_atoms 2 region box8
region box9 sphere 23.85599 25.339 29.7 25 units box
create_atoms 2 region box9
region box10 sphere 73.85599 25.339 29.7 25 units box
create_atoms 2 region box10
region box11 sphere 123.85599 25.339 29.7 25 units box
create_atoms 2 region box11
region box12 sphere 173.85599 25.339 29.7 25 units box
create_atoms 2 region box12
region box13 sphere 23.85599 75.339 29.7 25 units box
create_atoms 2 region box13
region box14 sphere 73.85599 75.339 29.7 25 units box
create_atoms 2 region box14
region box15 sphere 123.85599 75.339 29.7 25 units box
create_atoms 2 region box15
region box16 sphere 173.85599 75.339 29.7 25 units box
create_atoms 2 region box16
region left1 block -1.22802 0.22802 0 100.678 -1.7 1.7 units box
group left1 region left1
region right1 block 197.94 198.94 0 100.678 -1.7 1.7 units box
group right1 region right1
region left2 block 0.22802 197.94 0 0.5 -1.7 1.7 units box
group left2 region left2
region right2 block 0.22802 197.94 100.378 100.678 -1.7 1.7 units box
group right2 region right2
eam_potential
pair_style hybrid airebo 3 0 0 eam lj/cut 8.0625
pair_coeff * * airebo CH.airebo C NULL
pair_coeff 2 2 eam Cu_u3.eam
pair_coeff 1 2 lj/cut 0.01996 3.225
timestep 0.001
velocity all create 300.0 12345678 mom yes rot yes dist gaussian
velocity left1 create 0 12345678 mom yes rot yes dist gaussian
velocity left2 create 0 12345678 mom yes rot yes dist gaussian
velocity right1 create 0 12345678 mom yes rot yes dist gaussian
velocity right2 create 0 12345678 mom yes rot yes dist gaussian
fix 4 left1 setforce 0 0 0
fix 5 left2 setforce 0 0 0
fix 6 right1 setforce 0 0 0
fix 7 right2 setforce 0 0 0
thermo 100
thermo_style custom step etotal temp density pe ke
dump 1 all xyz 100 MDr.xyz
#process
fix 1 all nvt temp 300.0 300.0 0.1
run 200000
unfix 1
fix 2 all nvt temp 300.0 950 0.1
run 200000
unfix 2
fix 3 all nvt temp 950 950 0.1
run 200000
unfix 3
The fix 4,5,6,7 parts are placed in order to fix the four sides of the graphene with a very small width, because otherwise the graphene will stick to the nanoparticles and make the situation more difficult.
The code read_data data1.txt is related to the coordinates of carbon atoms and the dimensions of the simulation box, I have included a part of the data1 file below:
Carbon Positions in CNT Column1
7872 atoms
2 atom types
-15 240 xlo xhi
-15 120 ylo yhi
-75 75 zlo zhi
Masses
1 12.01
2 63.55
Atoms
1 1 0 0 0
2 1 -1.22802 0.709 0
3 1 -1.22802 2.127 0
4 1 0 2.836 0
…
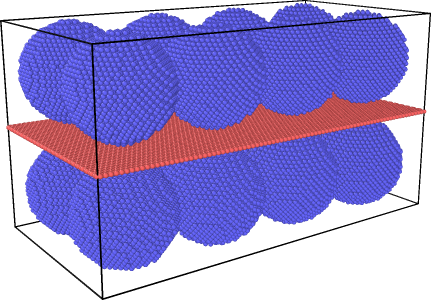

Does anyone have any suggestions to stop the copper nanoparticles from moving on the graphene?
Thank you for your attention dear ones