Dear User,
I am wondering if LAMMPS Colvars can specify the particular Volume in simulation system.(As shown in below figure),I would like to know whether it is possible to defined the region of V?
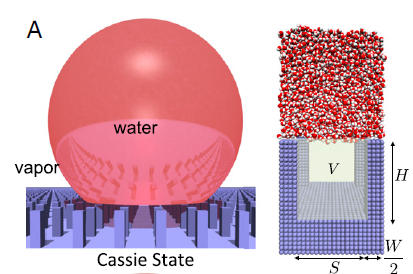
Looking forward to hearing from you.
Thank you very much in advance!
Best Regards
Liyi
Hi Liyi, you cannot specify a “region” in the sense of the LAMMPS commands that dynamically selects atoms, because the appearing and disappearing at the region’s boundaries would make the collective variable discontinuous. You can, however, define a “group” with the atoms of interest and for example study the PMF of displacement along the vertical axis of your problem. (Beware that the computation may expensive, as it is not fully parallelized: test this for your system).
For your convenience, you can select atoms using Colvars commands or LAMMPS commands and converting back and forth with:
http://lammps.sandia.gov/doc/group2ndx.html
A variable you may want to look at is distanceZ, which supports umbrella sampling, metadynamics, ABF, etc.
Giacomo
Dear User,
Thank you very much for your promptly reply.
The collective variable I want to study is the number of water molecules in the specific region V.Sorry.I would like to make your points cleared. You mean I can only define some interested atom in the entire system instead of the atoms on the specified Volume V?
Thank you very much for your help.
Best regards
Liyi
[

liyi bai
邮箱:liyibai2011@…24…
](https://maas.mail.163.com/dashi-web-extend/html/proSignature.html?iconUrl=http%3A%2F%2Fnos.netease.com%2Fmail-online%2F8931148f878b801829a41f7b4b58591c.jpg&name=liyi%20bai&uid=example%40163.com&ftlId=1&items=["邮箱:liyibai2011%40gmail.com"])
签名由 网易邮箱大师 定制
Dear User,
I have a name, thank you.
Thank you very much for your promptly reply.
The collective variable I want to study is the number of water molecules
in the specific region V.Sorry.I would like to make your points cleared.
You mean I can only define some interested atom in the entire system
instead of the atoms on the specified Volume V?
You must define a function of all the atoms in the system, the values of
which are going to be non-zero only for the atoms that you are interested
in. You could also try that, although what I suggested (using the
center-of-mass-displacement of the droplet is somewhat simpler.
A general suggestion that I have is to load a trajectory into VMD or use
the rerun command to test in advance how the chosen variable would behave.
Giacomo

![https://maas.mail.163.com/dashi-web-extend/html/proSignature.html?iconUrl=http%3A%2F%2Fnos.netease.com%2Fmail-online%2F8931148f878b801829a41f7b4b58591c.jpg&name=liyi%20bai&uid=example%40163.com&ftlId=1&items=["邮箱:liyibai2011%40gmail.com"]](https://maas.mail.163.com/dashi-web-extend/html/proSignature.html?iconUrl=http%3A%2F%2Fnos.netease.com%2Fmail-online%2F8931148f878b801829a41f7b4b58591c.jpg&name=liyi%20bai&uid=example%40163.com&ftlId=1&items=%5B%22%E9%82%AE%E7%AE%B1%EF%BC%9Aliyibai2011%40gmail.com%22%5D){kind=link}