Dear Lammps Users,as the picture when i running lammps , the “job aborted” prompt appears, I don’t know how to modify or which files should be checked out to solve this problem, I hope to get your help! thanks!
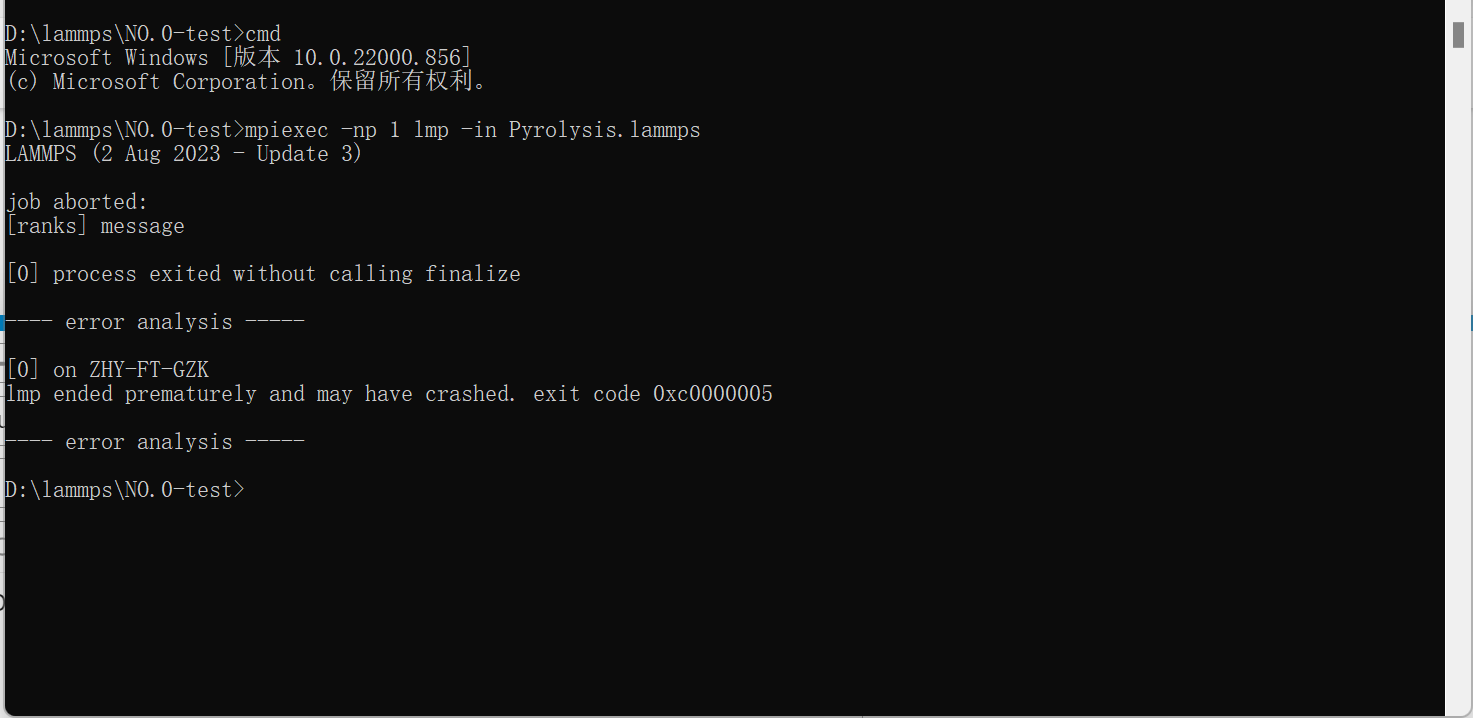
Likely there is an error in your Pyrolysis.lammps input file.
The actual LAMMPS error message seems to be hidden due to Windows/MPI buffering of standard output. You can try adding the flags -echo screen -nb to your command line to have more detailed logging to the screen and to turn of buffering.
thank you Mr.`Kohlmeyer!
Mr.Kohlmeyer
Thank you for your advice,lammps can already be run successfully.However, an error
ERROR: Lost atoms: original 450 current 449 (src/thermo.cpp:488)
occurred during the operation.How should I get a more detailed information about the error and solve this error?Hope to get your reply.
Pyrolysis.lammps (2.8 KB)
ffield.reax (22.2 KB)
HCFO1233ydZ.data (23.1 KB)
First you need to do your due diligence and research this error. It is one of the most reported errors and thus there are many discussions in the archives about possible causes (there can be multiple) and remedies. Since you are running ReaxFF for pyrolysis, i.e. at high temperature, it is most likely that your timestep is too large. ReaxFF needs a much shorter timestep than other force fields.
Your input has
neigh_modify every 500 delay 0 check no
That is a VERY bad idea to set reneighboring to be this infrequent and without checking.
The savings of doing neighbor list builds are miniscule already when using “every 10” since you are already saving 90% of the cost. Specifically for an “expensive” force field like ReaxFF the cost is elsewhere, e.g. with the charge equilibration and the design of the force field in general.
Thank you Mr.Kohlmeyer!
I have benefited a lot from your reply.I will modify
neigh_modify every 500 delay 0 check no
and try again.
I am a chemical engineer, and I am learning to use lammps. Now I mainly check the manual and log in some online communities to conduct lammps error research. Like MATSCI, are there any other ways to recommend it? Or do you have any other suggestions on my way to learn lammps?
Look forward to your response.
You need to start with getting a text book on MD simulations. Some suggestions are here: Books about Molecular Dynamics generally or LAMMPS specifically. Learning some statistical mechanics and statistical thermodynamics does not hurt either.
Then you need to get a tutor or something equivalent. MD simulations are as much craft as they are a science and the MD simulation craft you can only properly learn from an MD practitioner. The best way is to work with a group that has that experience for a while. If there is nobody local, you may need to visit a such a group for a few weeks.
Thanks for your advice!