Dears LAMMPS community,
Summary: I am running a simulation to replicate a previous simulation work has been done using different force field (DOI: https://doi.org/10.11113/jt.v79.11322). They used COMPASS force field while I am using OPLSaa. The idea is to compare with their experimental and simulation result. I am getting some reasonable results however others are not making sense for me.
I run the equilibration stages using
1- NVE then NPT I took the box size (density and temperature were stable but pressure fluctuate ±1000 please see the summary below)
2- then use NVT (I was hoping to minimize the pressure fluctuation but it didn’t >> run it for 10K step just to make sure)
3- production stage: NVT and calculate the RDFs
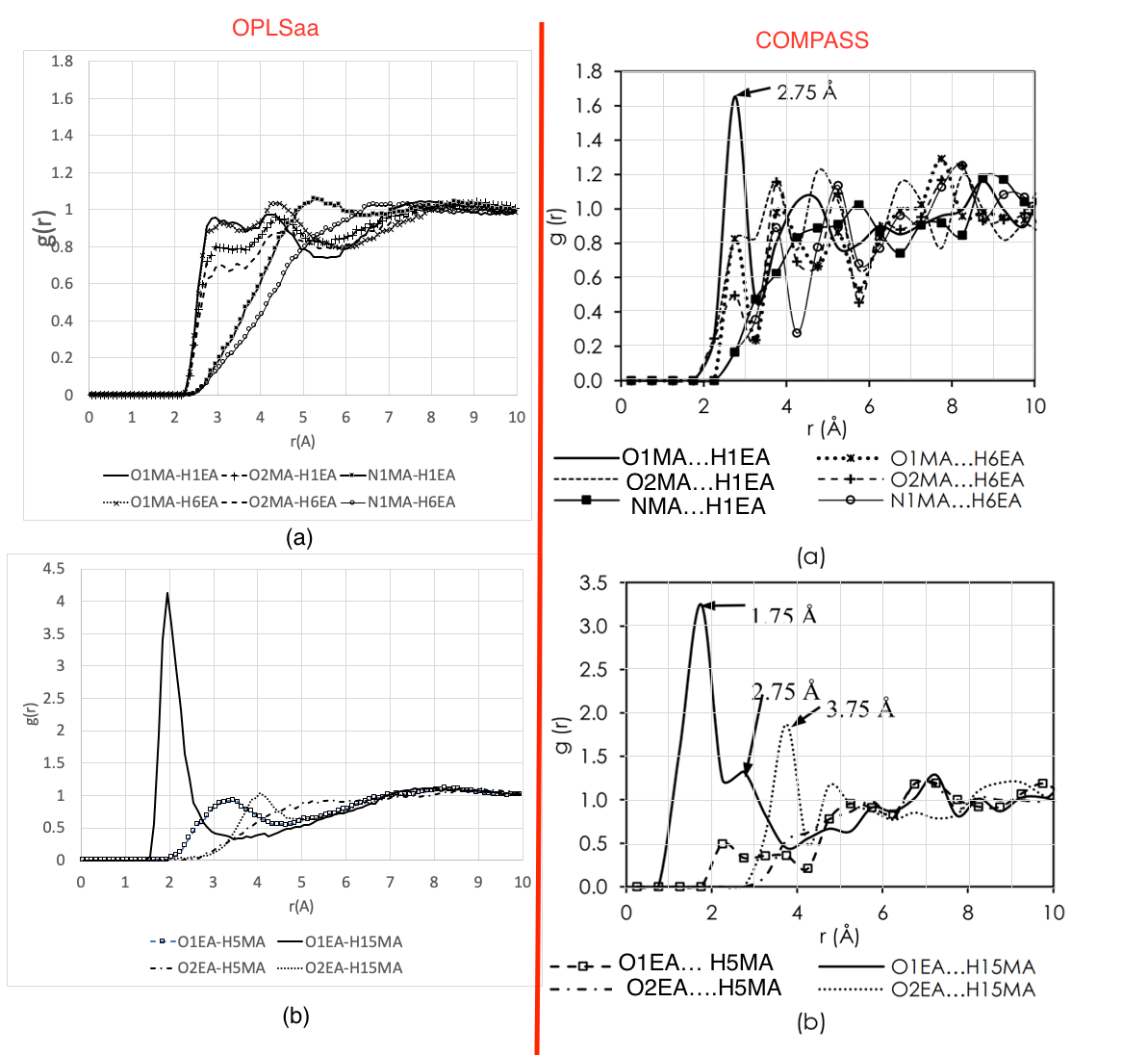
I was able to get density (0.934g/cm3) close to the experimental density (0.94g/cm3). However, the RDF is not in agreement (i attached a picture showing the RDFs from my simulation OPLSaa left column vs COMPASS right side).
I checked my script and the atom assignments and the potentials (which is generated using Moltemplate). but couldn’t figure out and kind of get lost. I would be very thankful to give me advice about where and what to check is it a script? is it force field limitation? is this related to the pressure fluctuation? Or other aspects I am not taking into account?
Summary and part of the script is written bellow
Many thanks,
Moulham
PhD student at university of Strathclyde
summary