Hi,
I’m want to simulate graphene with Tersoff potential, but this error comes up and I don"t know why or what can I do to fix it. Please help.
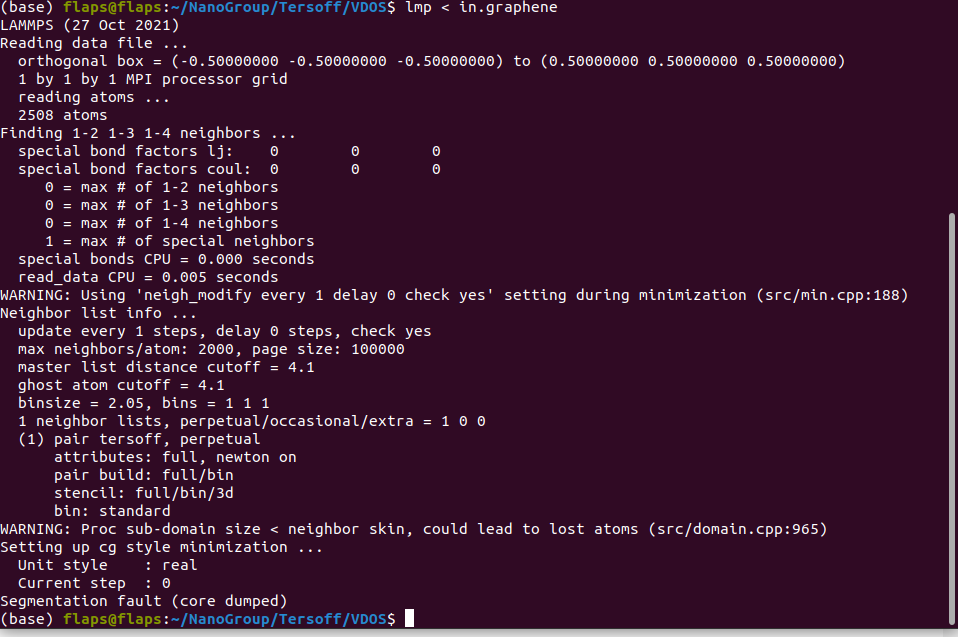
Here is my code and input.
in.graphene (877 Bytes)
Ctersoff1.dat (428 Bytes)
pos.data (142.1 KB)
Hi,
I’m want to simulate graphene with Tersoff potential, but this error comes up and I don"t know why or what can I do to fix it. Please help.
Hello,
You have 2508 atoms in a 1 Angstrom³ box…
Simon
Omg, such a basic error.
I forgot that VMD gives the units in nm. Thank you so much.
That is not correct. VMD users Angstrom as units.
Instead, what must have happened is that you imported coordinates from a file that does not store box information and hence your TopoTools version used its default of 1x1x1 angstrom.
Your v1.7 version of TopoTools is outdated: Releases · akohlmey/topotools · GitHub
More recent versions will stop with an error if you try to write out a system without setting box dimensions explicitly.