Trying to set different styles of interaction with the following code
pair_style hybrid gran/hooke/history 1e-4 NULL 1e-5 NULL 0.0 1 lj/cut 2.5e-6
pair_coeff * * gran/hooke/history
pair_coeff 1 3 lj/cut 1.0 1.0 2.5e-6
gives me a segmentation fault.
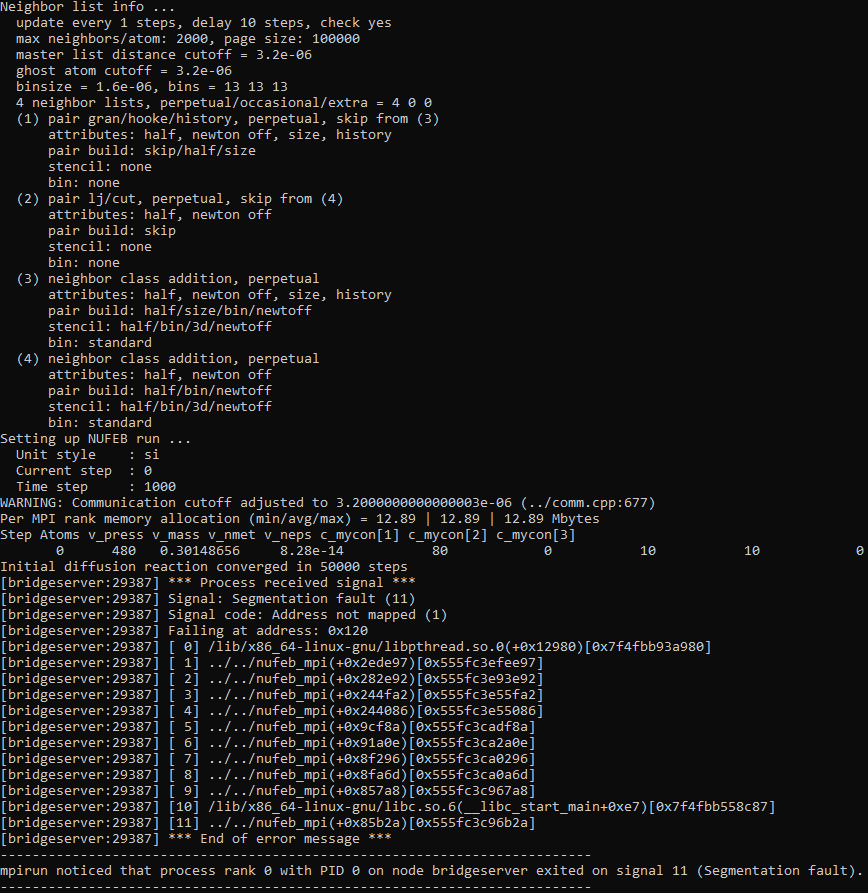
This is a NUFEB simulation, where I am trying to simulate different interactions between cells (atoms type 1) and ganular activated carbon (atoms type 3).
# NUFEB simulation with methanogens and GAC floor
units si # using si units
atom_style coccus # using nufeb atom style
# map array: find atoms using indices sort 1000 5.0e-6: sort every 1000 steps with 5.0e-6 binsize
atom_modify map array sort 1000 1e-6
# periodic boundaries in x and y fixed boundary in z
boundary ff ff ff
# forces between local and ghost atoms are computed in each processor without communication
newton off
processors * * * # processor grid
# communicate velocities for ghost atoms
comm_modify vel yes
# guarantee that enough atoms are communicated to correctly compute
comm_modify cutoff 2e-6
read_data gac_pure_culture.in
# Shift the lattice grid 0.5 unit so that atom can be created in the center of the grid
lattice sc 1e-6 origin 0.5 0.5 0.5
region reg block 0 20 0 20 4 5
create_atoms 1 random 80 31324 reg
# create the GAC floor - this is influenced by the lattice command
region gac_floor block 0 20 0 20 0 1
create_atoms 3 region gac_floor
region outside_gac_floor block 0 20 0 20 0 1
set type 1 density 150
set type 1 diameter 1.3e-6
set type 1 mass 1.725e-16
set type 1 outer_diameter 1.3e-6
set type 1 outer_density 30
# define attributes for type 3, set gac diameter to 1e-6, make it consistent with lattice size
set type 3 density 150
set type 3 mass 1.725e-16
set type 3 diameter 1e-6
set type 3 outer_diameter 1e-6
set type 3 outer_density 30
# set the groups
group met type 1
group eps type 2
group non-gac type 1 2
group gac type 3
# setting neighbour skin distance and style
neighbor 7e-7 bin
# rebuild neighbour list if any atom had moved more than half the skin distance
neigh_modify check yes
# select grid style
grid_style nufeb/chemostat 3 h2 co2 ch4 2e-6
# test grid comm
#run_style test/comm_grid
#run 0
# set substrates initial concentration
grid_modify set h2 nn nn nn 10 10
grid_modify set co2 nn nn nn 10 10
grid_modify set ch4 nn nn nn 0 0
# define pair styles
pair_style hybrid gran/hooke/history 1e-4 NULL 1e-5 NULL 0.0 1 lj/cut 2.5e-6
pair_coeff * * gran/hooke/history
pair_coeff 1 3 lj/cut 1.0 1.0 2.5e-6
# NVE integration with maximum distance limit
# only update position for non-gac atoms
fix nve non-gac nve/limit 1e-8
# monod reaction fixes
fix monod_met met nufeb/growth/methanogen h2 6e-6 co2 2e-4 ch4 growth 1.92e-4 yield 1.6 decay 0 epsyield 1 epsdens 30
# diffusion reaction fixes
fix diff_sub all nufeb/diffusion_reaction h2 4.5e-9
fix diff_co2 all nufeb/diffusion_reaction co2 1.186111e-9
fix diff_ch4 all nufeb/diffusion_reaction ch4 1.488889e-9
# biological model fixes
fix div met nufeb/division/limited 1.36e-6 30 1234 region_blocked gac_floor
fix eps_ext met nufeb/eps_extract 2 eps 1.3 30 5678
# mechanical model fixes
fix wall all wall/gran hooke/history 1e-3 NULL 1e-4 NULL 0 0 zplane 0.0 8e-5
fix eps_adh all nufeb/adhesion/eps eps 1e-6
fix vis all viscous 1e-5
# fix ID atomsID nufeb/shear shearRate viscosity +x (or -x or +y or -y)
fix s1 non-gac nufeb/shear 0.2 0.001 +x
# pressure computation
compute vol all nufeb/volume
compute ke all ke
variable one equal 1.0
compute press all pressure NULL pair vol v_one
variable press equal "(c_ke + c_press) / (3.0 * c_vol)"
variable mass equal "mass(all)"
variable nmet equal "count(met)"
variable neps equal "count(eps)"
compute mycon all nufeb/ave_conc
variable ch4 equal c_mycon[2]
# file output
shell mkdir vtk_gac_sheet
dump 1 all vtk 10 vtk_gac_sheet/dump*.vtu id type diameter
dump 2 all grid/vtk 10 vtk_gac_sheet/dump_%_*.vti con
# thermo output
thermo_style custom step atoms v_press v_mass v_nmet v_neps c_mycon[*]
thermo 1
thermo_modify lost ignore flush yes
# issue run command
run_style nufeb diffdt 1e-4 difftol 1e-12 pairdt 1e-2 pairtol 1 pairmax 1000 diffmax 50000
timestep 1000
run 150
NUFEB uses an outdated version of LAMMPS, so I cannot update LAMMPS to a newer one. Could just the older version be the reason for this problem? Or should I specify some other parameter that LAMMPS is not finding, just reporting that it can’t find its value with “address not mapped”?