Dear All
I want to use MC/MD simulations for a multi-elemental alloy. For this, I want to incorporate SGCMC for lammps.
Some of the codes are shown here.
units metal
atom_style atomic
boundary p p p
dimension 3
neighbor 3.0 bin
read_data final.lmp
variable temperature equal 500.0
timestep 0.0025
compute Tall all temp
compute_modify Tall dynamic/dof yes
compute Pall all pressure Tall
compute_modify Pall dynamic/dof yes
compute ke all ke/atom
compute pe all pe/atom
pair_style eam/alloy
pair_coeff * * FeCrNi_d.eam.alloy Fe Cr Ni
velocity all create 500.0 428459 dist gaussian
fix 2 all npt temp 500.0 500.0 0.1 aniso 0.0 0.0 1.5
fix_modify 2 temp Tall press Pall
fix mc all sgcmc 20 0.25 500.0 -0.3 -0.1 randseed 324234 variance 1e3 0.032 0.0132
thermo_modify lost ignore
run 5000000
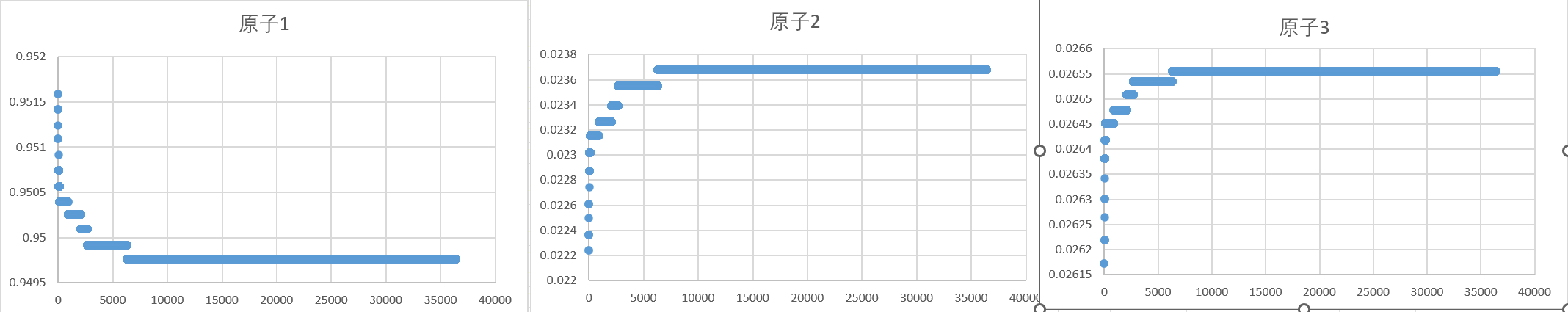
Unfortunately, the final calculated concentrations are 0.9497, 0.0237, 0.0266. Why is this situation?
And the statement on the official website about sgcmc using other types of potential functions is " If other potentials are to be used it is strongly recommended to modify and optimize the existing generic potential functions for this purpose. “. I want to use hybrid potential, which includes eam/alloy and eam/fs, but I do not know how to modify and optimize the existing generic potential functions.
Thanks a lot!
Dora
Please study Please Read This First: Guidelines and Suggestions for posting LAMMPS questions and follow the guidelines presented there.
Your post violates several of those guidelines which makes it extremely difficult to provide any meaningful response.
I am sorry for the issues mentioned above. I will improve them and publish them again. Thank you for your reply very much.
Dear akohlmey, I have completed the modification of the question. Please check if it is correct. Thank you.
It is a little bit better, but there are still issues where you do not follow the guidelines, most importantly
what is your LAMMPS version and what is the platform you are running on?
This is difficult to say without being able to experiment and from just looking at a few lines of input. What is the evolution of the concentrations? How many atoms has your system? … and from that, please compute how many atoms would have to be in the system to represent the desired values.
Why that command? How many atoms do you lose over time? Do you want atoms to leave the system? Your system looks like a regular bulk system, so there should not be any lost atoms and thus the command should be removed. If you cannot run the simulation to completion without, you have to figure out why this is the case. Lost atoms for a bulk system with periodic boundaries usually are an indication of bad simulation settings, bad (initial?) geometry, or bad force field parameters.
Which website exactly?
That is a very bad idea. Those two potentials have the same computation, but just have the parameters formatted differently. You should convert or recreate the potential file to the same format. Using hybrid potential with EAM is particularly bad, since you will have a bad model as the embedding function cannot be computed between atom associated with the two different pair styles. You can easily make a test and just use two atom types but map both to the atom types to same element in pair_coeff command and then compare the results to a simulation where you define the same pair style twice and then map each atom type to a different pair style instance. Do you get the same results in that setup? Your statement seems to imply that you would. I would argue against it. There are many discussions on the subject in the archives of this forum, as (ab-)using of the hybrid pair style is a common theme.
There is a whole section in the LAMMPS manual dedicated to modifying and extending LAMMPS. You just need to start reading: 3. Modifying & extending LAMMPS — LAMMPS documentation
Thanks for your reply very much. My LAMMPS version is 28 Mar 2023. I run the programs on Linux. But I have a question, will different versions or platforms lead to different computational results?
I checked the atomic concentration near the end of the run.
My system have 300000 atoms. The numbers of atoms I need are 286440, 9600, 3960.
The system lost 9 atoms in total at the end of the run. The step size is 0.0025 ps, running a total of 5000000 steps. By the way, I have no idea about the steps required.
The website is fix sgcmc command — LAMMPS documentation
I’m guessing if the potential function caused the calculation error, so I want to use the hybrid potential. I will try to recreate the potential file to the same format.
Thank you very much, Your reply has greatly inspired me.
It does not have to. This is mostly to check for bugs and bugfixes. Some people use very old versions of LAMMPS (>2years old), then the risk increases. It can make a difference which compiler you use. Certain versions of the Intel compilers are particularly liable to miscompile some functions when aggressive optimization is requested, but the same can happen for other compilers, too. You most certainly won’t always get bitwise identical results. Those can change with the number of processors used or the level of compiler optimization, since we are using floating point mach and any process that changes the order in which some numbers are summed up can lead to differences in rounding. Please see the following blog posts: Floating Point Demystified, Part 1, Floating Point Demystified, Part 2: Why Doesn't 0.1 + 0.2 == 0.3?, and http://floating-point-gui.de/
This is very worrisome. You need to figure out where this comes from.
My suggestion is: set up a test system simulation with 1/10th the atoms so that you can do more timesteps faster. Change your input so that you have thermo_modify lost warn and thus get a note immediately when a lost atom is detected. Then see, if the lost atoms still happen. If yes, then test without fix sgcmc. If you still have lost atoms then, I would suggest you try reducing the timestep to 1fs, and if that does not suffice to 0.25 fs. If reducing the timestep helps, then re-add fix sgcmc and check again.
Your choice of the number of steps is rather conservative. Your graphs show that the simulation already has stabilized. Unfortunately, there are no axis labels and the titles are not in english.
It is always a bad idea to guess. What this paragraph is referring to is that for optimal performance, the fix sgcmc command has some optimizations that can only work for EAM style pair styles. Looking at the code in question, that should be revised to also support all pair styles that are not manybody potentials and do not use long-range electrostatics. Otherwise, pair styles need to be modified, but those modifications are difficult to get right. When merging the standalone version we removed several such modifications, since they were not implemented cleanly and had other issues.
Bottom line, switching to a different potential file with different parameters will not have an impact on this, for as long as you stay within EAM potentials. Otherwise, the fix will use a much slower but generic algorithm. In fact, if you cannot find what is the problem causing the lost atoms, we can discuss how to change the fix source code so that the slow method is always used and then we can confirm if the discrepancy is due to this optimization.
I would also strongly advise to study the publication Sadigh1 to understand how well this method based on using target concentrations can work, and whether the discrepancy you see is consistent with the tests done by the authors of the paper or not.
I have got what you have said here, and I will make the modifications. Thank you very much.