Dear all,
I am working with a system that contains X number of polymer chains and Y number of water molecules.
I wanted to insert an additional 1000 water molecules into the simulation box. To do this, I first changed the box size to the closest (larger) integer value for each dimension (without remapping). I then used ‘create_atoms’ to insert all new water molecules randomly in the box - using a molecule template. I deleted all water molecules within a 1 Angstrom cut-off of any other atom, repeatedly using ‘create_atoms’ until all waters were inserted with no further deletions.
This seemed at first successful. I minimised the system to a low potential energy and force tolerance and a further simulation at 300K with fix NPT ran without error (or warning). The problems started when heating to 400K. I get the error ‘lost shake atoms A B C on proc X’ semi-consistently somewhere in the middle of the simulation (i.e., after 75000 1fs timesteps).
I have included two snapshots of the simulation box (each from a trajectory file) below. The first is of the system before additional waters are inserted and the second is after. Both have been wrapped by residue/molecule and I am just showing water molecules for clarity here.
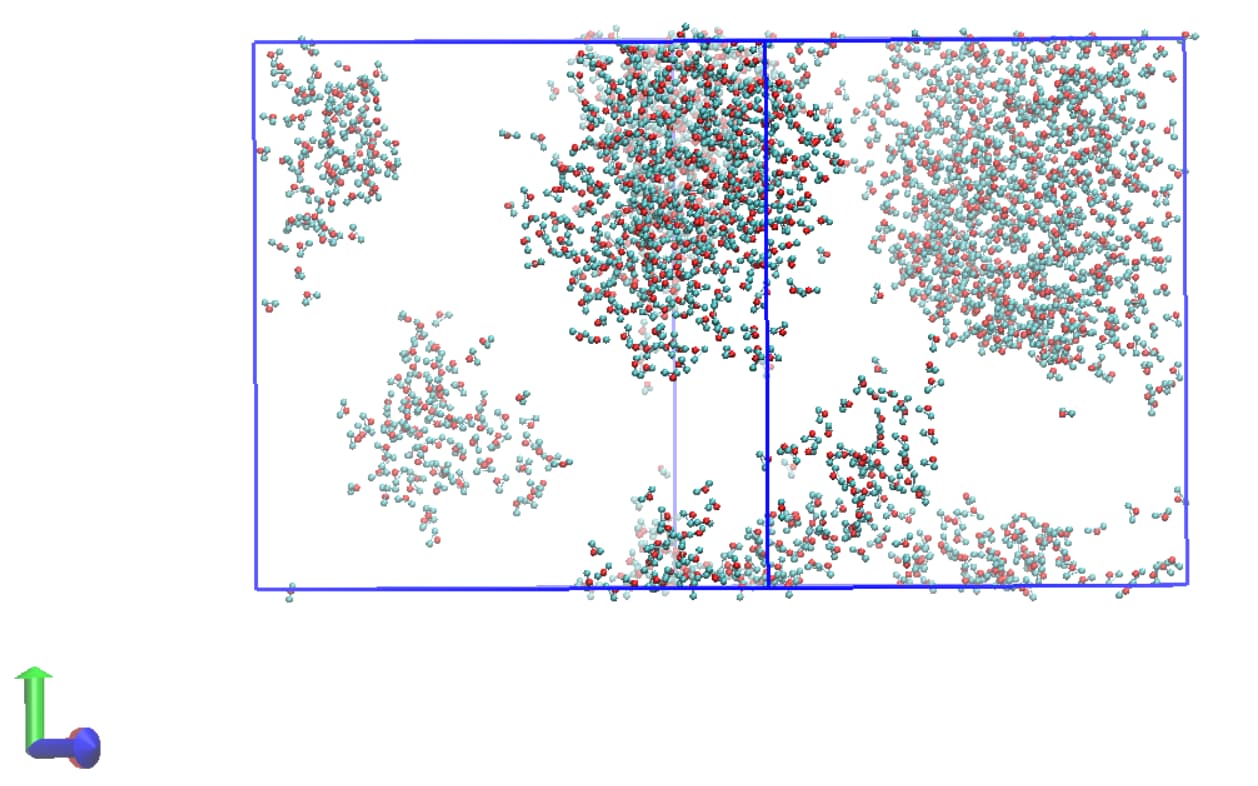
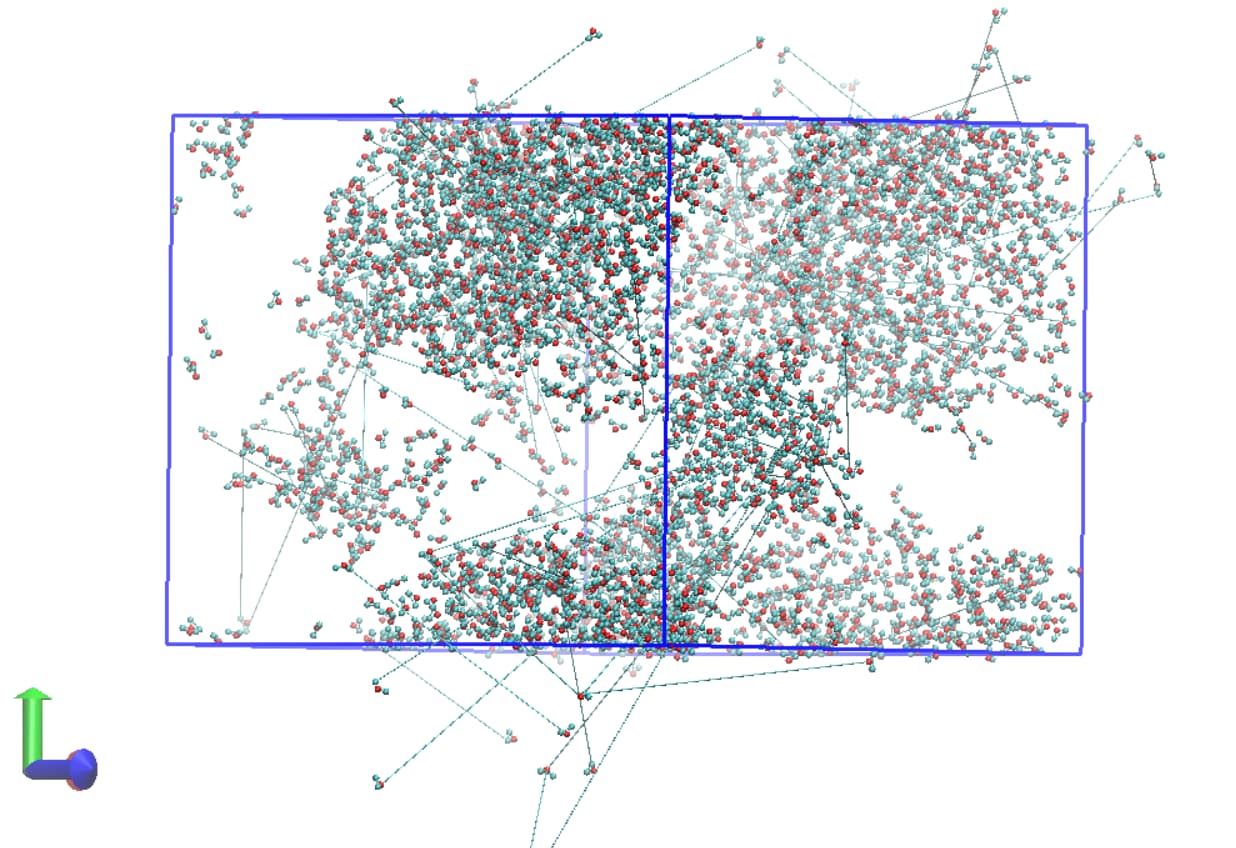
It’s clear that there is some issue with incorrect image flags for atoms water molecules at the periodic boundary leading to the appearance of bonding between different molecules when the trajectory is visualised. It’s worth noting that VMD reads the bonding for the trajectory files incorrectly (assigning a bond between water hydrogens for example) and that the topology file itself shows the correct bonding.
Would greatly appreciate any help/advice with this.
Thanks