I am very new to LAMMPS trying to run some simple NEMD cases with the code (so sorry if the questions are basic). I am trying to simulate a system in shear flow using the nemd commands shown below. The system consists of multiple particles which can interact with each other. What I observed is that when I visualize the output in VMD, the box deforms in one direction while the particles are moving in the opposite direction. This does not make sense to me at least visually. I was wondering if I am doing something wrong or there exists an explanation for this. here are the main lines of the script I am running:
units lj
atom_style atomic
boundary p p p
read_data initial_configuration.txt (the data includes initial positions and velocities + dimensions of orthogonal box)
change_box all x delta 0 20 boundary p p p triclinic
fix 2 all langevin 1.0 1.0 1.0 ${seed} #(may comment sometimes)
fix 1 all nvt/sllod temp 1.0 1.0 1.0
fix 3 all deform 1 xy erate 0.01 remap v
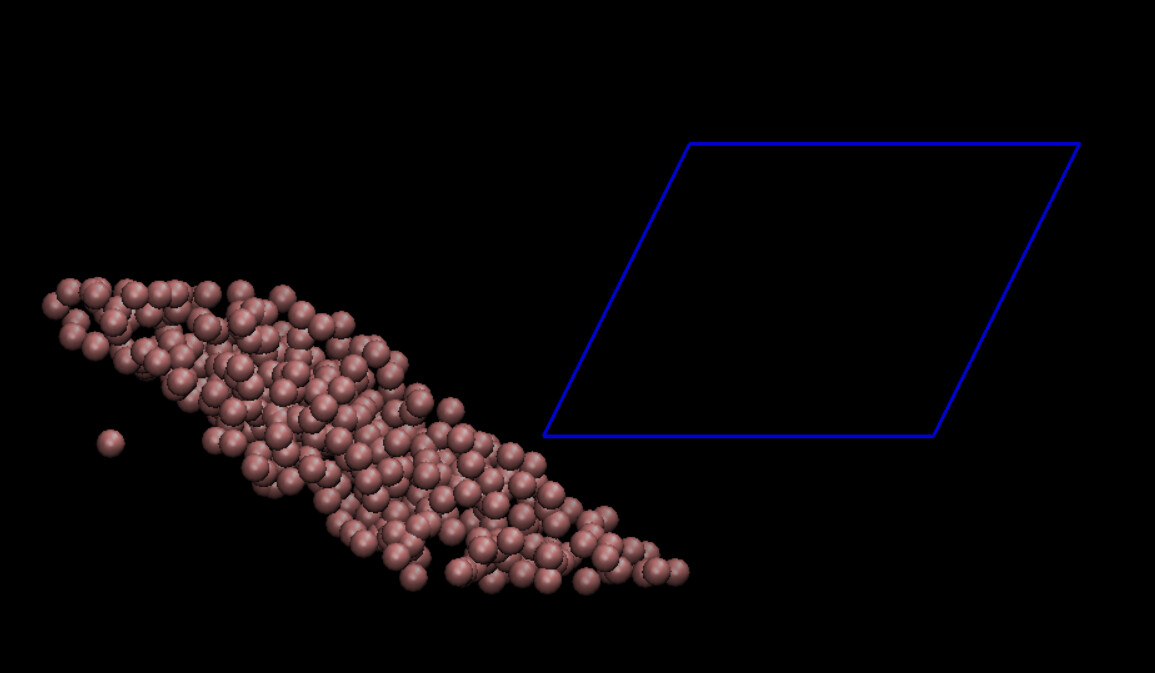
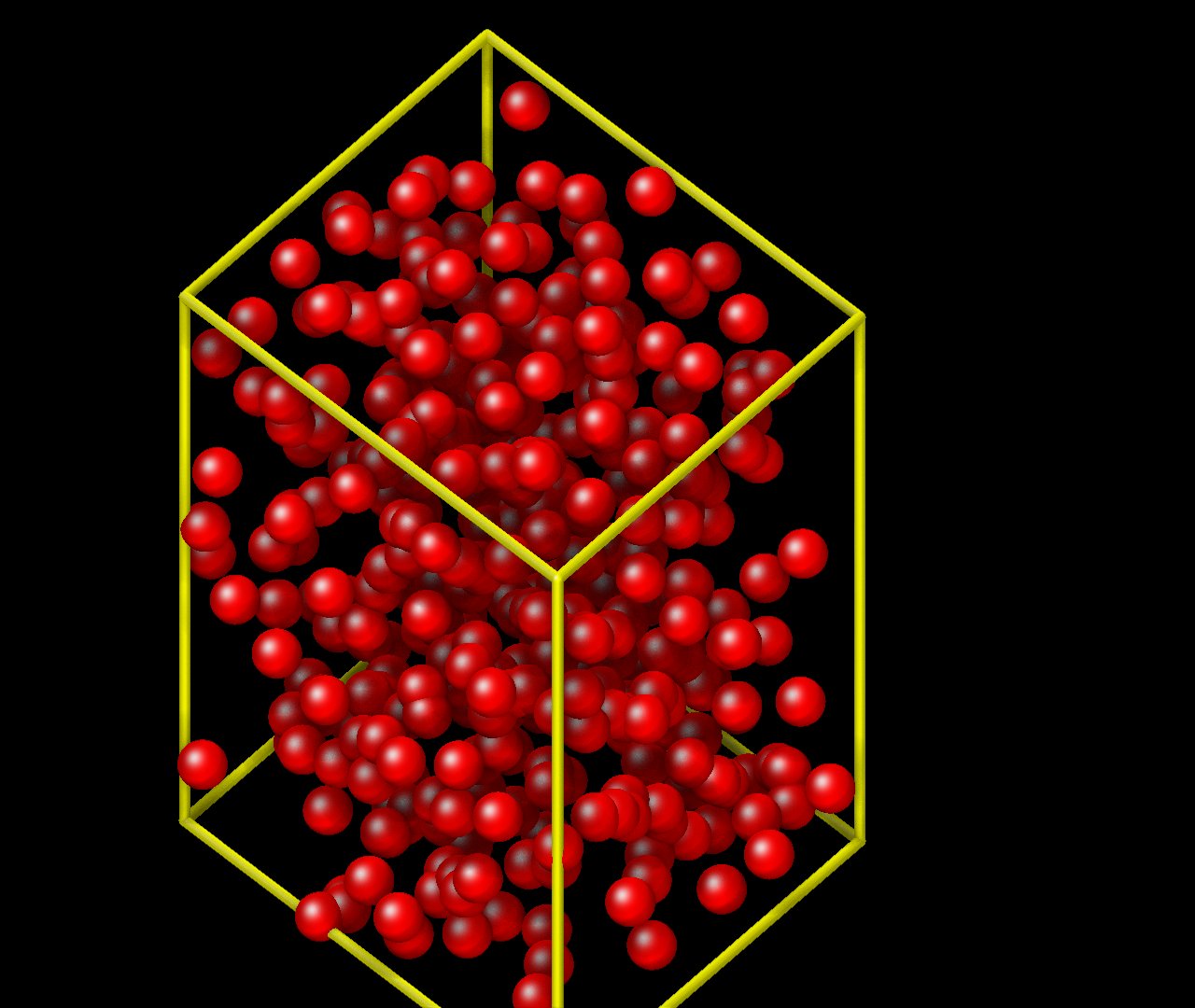
Here is an image of what I get in VMD after few timesteps:
Another minor questions is that although the initial configuration puts all atoms in the box, whenever I visualize in VMD it seems that only 1/4 of the particles are inside the visualized box. Is there an explanation for it?
Please report which versions of LAMMPS and VMD you are using and it would also be great if you could provide the complete input deck (best create a compressed tar archive, put it on dropbox, google drive, one drive, github or similar and provide a link to it) so we can try to reproduce your observations and review all settings and input.
Thanks for the quick reply. The LAMMPs I am using is on hpc and the 22 Aug 2018 version. VMD is the latest 1.9.4 windows 64bit version (2020). The script and initial configuration files are located here:
Thanks for the files. The problem is that you are looking at “unwrapped” coordinates.
If you would write out the coordinates without the image flags (just x y z) and visualize those, you should see that they do follow the box deformation, but also that the box will “snap back” after it has reached 50% tilt and that the atoms will follow the induced flow.
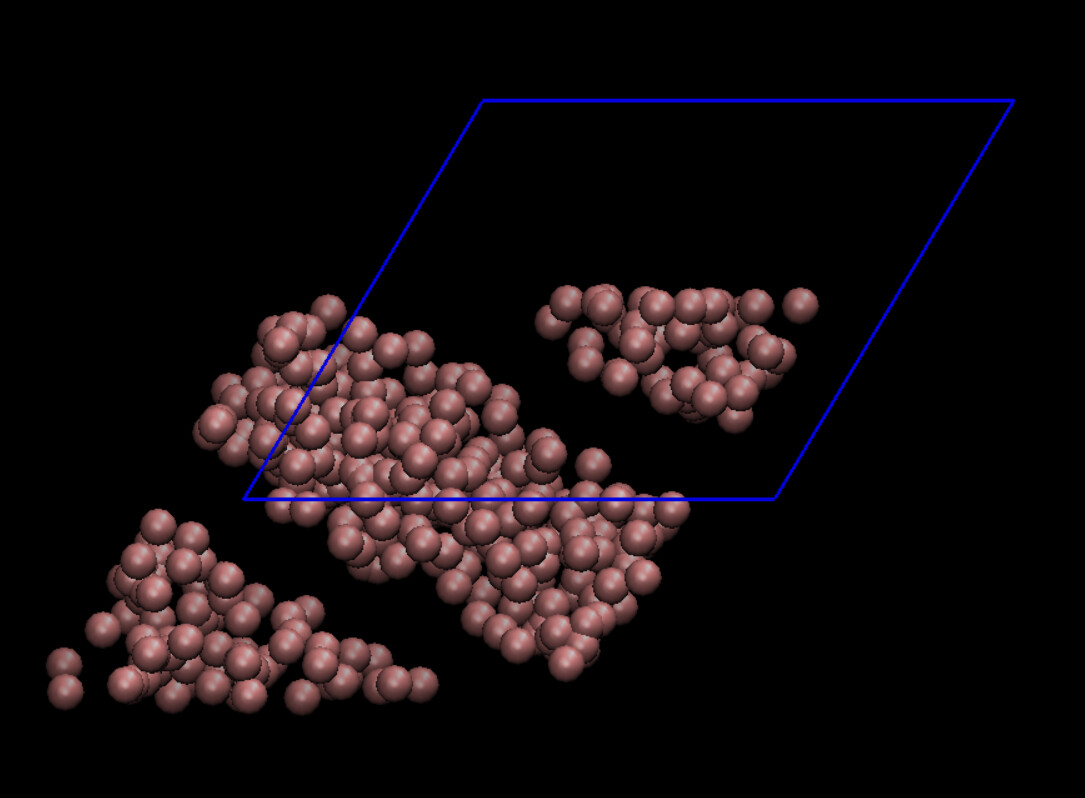
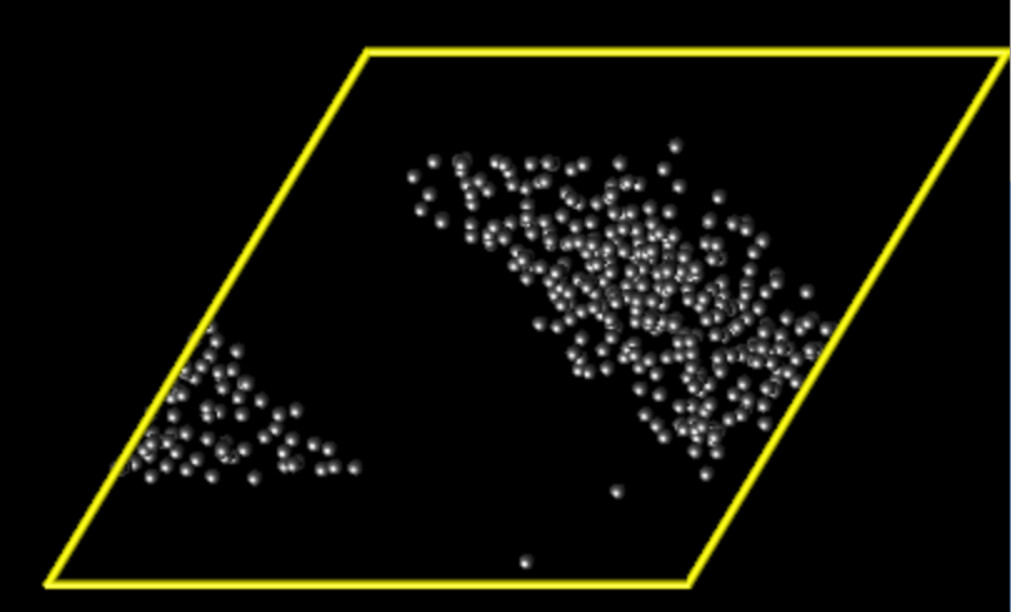
Thanks so much for your help. The deformation looks fine now and I have attached as image.
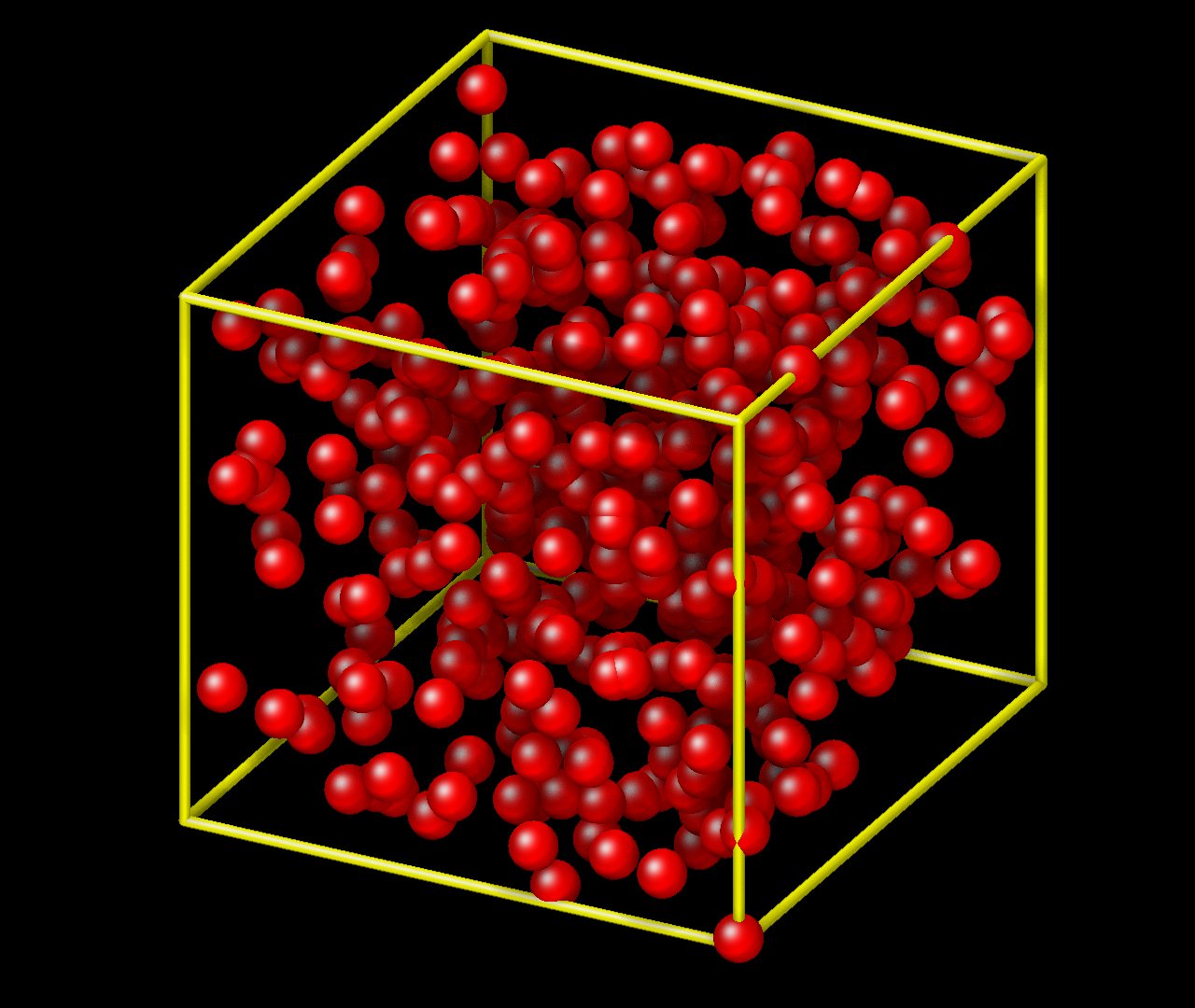
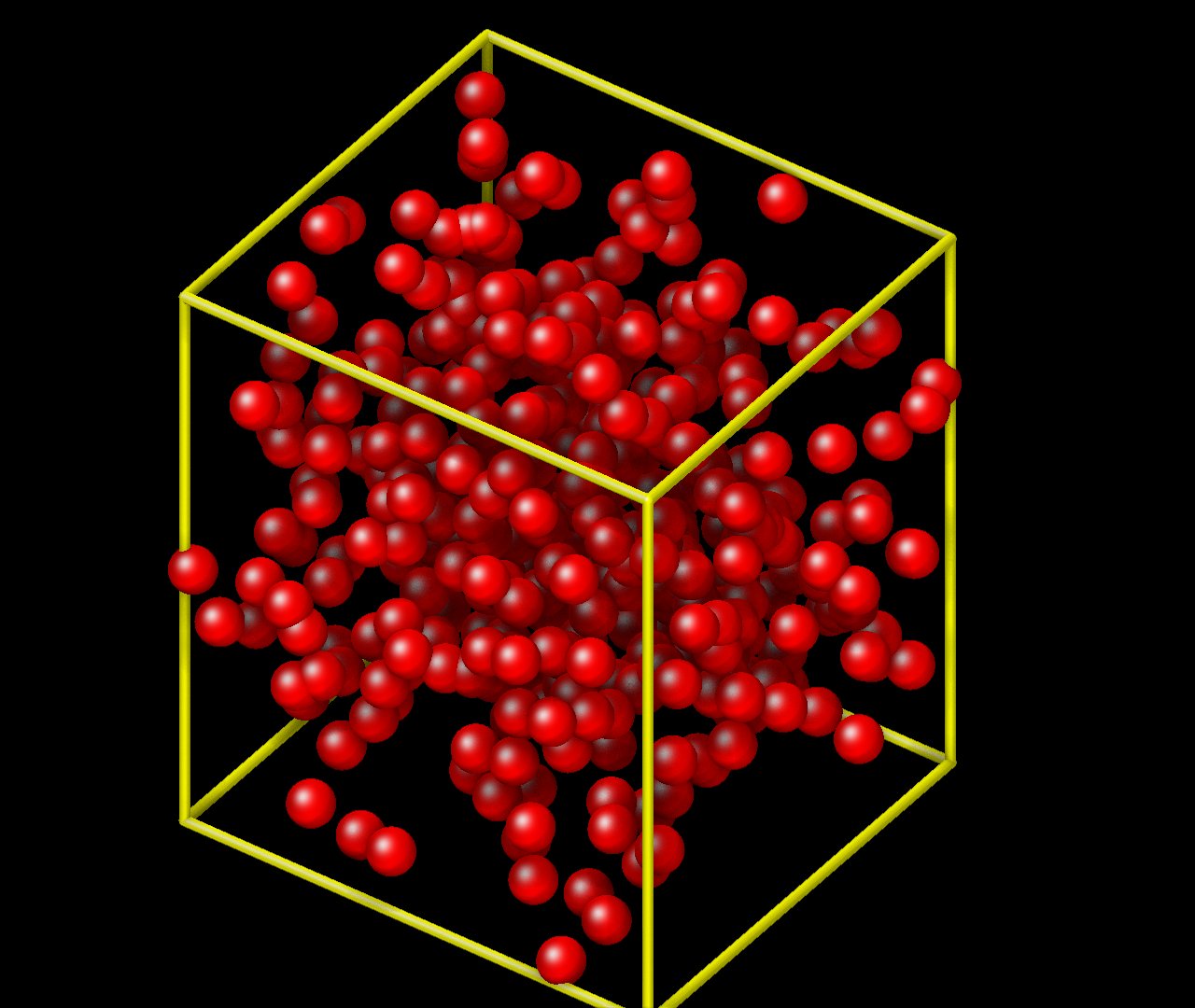
Regarding the bead positions being out of box, is there also a problem with the output coordinates. The allocated bead positions initially are all within the xlo and xhi bounds of the box but visualizing it, it seems like only a portion of them are within the representative box at lease when I use VMD.
This is a VMD issue. VMD internally only stores the box lengths and angles, but not the location of the origin. You’ll have to tell the pbc box command where to place the origin. This issue can be avoided when creating images with dump image in LAMMPS directly:
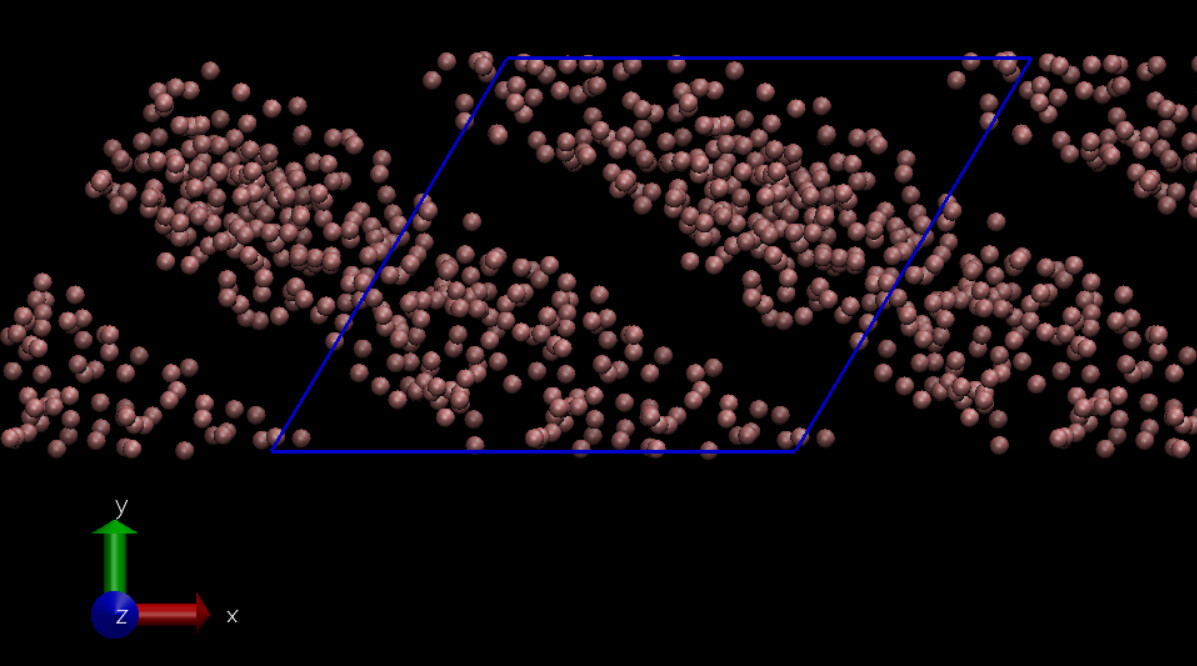
So I used the dump images ppm and visualized the box and particle deformations using LAMMPS output (using also only x y z and not the unwrapped ones). It again looks like the deformation is happening in the opposite direction (Image attached). Going to VMD and adding the periodic images, the same thing happens there as well (image attached) so the image I previously sent is a bit misleading. I also checked the coordinates in the dump file and what I expect is to see a particle in positive y coordinates initially, moves in the positive x axis as the simulation goes but that is not the case.
I don’t understand that LAMMPS image. What is the time=0 image, e.g. before the box starts deforming? I assume your system is periodic in y ? For examples of 2d system where this works, see the examples/VISCOSITY/in.nemd.2d script and uncomment the dump image or dump movie commands. Maybe compare that script to yours? Movies for this script are the last ones on the LAMMPS website Movies page.
It’s certainly possible in LAMMPS to shear a fluid so that the fluid does not move with the deforming box. But it’s not possible to have the atoms be far outside the box.