Dear Shan,
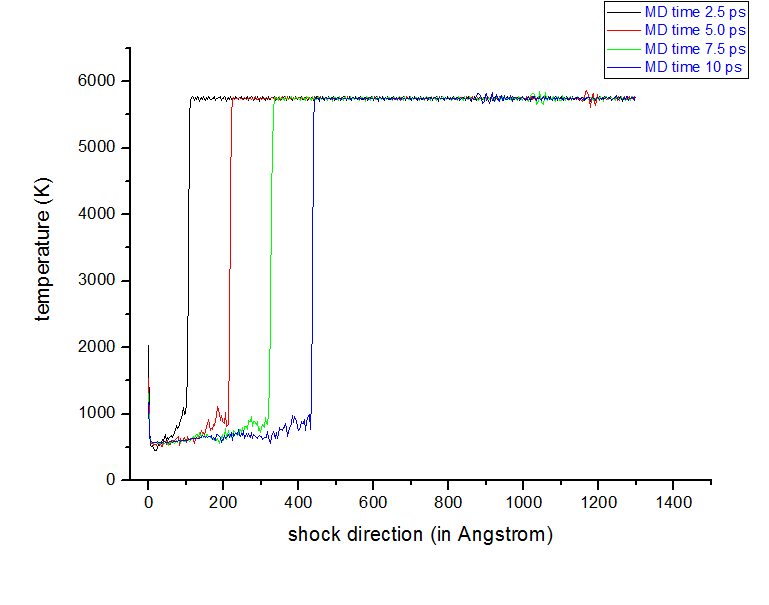
it was a mistake while I was pasting my script, I am furnishing the full script here…please see, BTW regarding electronic contribution of temperature, you are quite correct but to get such high temperature been possible for shock experiment ? I mean at 10000 K copper copper should be in vapor state, how can I justify this. I have gone through a lots of literature but most of them are not mentioning the temperature.
NEMD Shock experiment with a copper bulk
System dimensiion 400100100 unit cells in X,Y and Z direction
Method: Single Impact Shock
Potential: EAM cu Foiels et al
#-----------------------Initialization-------------------------------------#
variable kB equal 1.3806504e-23 # [J/K] Boltzmann
variable Na equal 6.02e23 # Avogardo number
variable pconv equal 0.0001 # bar to GPa
variable vconv equal 0.100 # A/ps to Km/sec
variable wallleft equal -8.05858552526837698e-03 # in length unit
variable wallright equal 1.4462248969819657e+03 # in length unit
variable SHOCKVEL equal 1.5 # km/s
variable Init_Vol equal 10455.6233191.0e-24 # Cm3
variable Init_Leng equal 144.614430415 # in Angstrom
variable vel equal ${SHOCKVEL}-10 # A/ps
#-----------------------System Variable Initialization ---------------------#
units metal
atom_style atomic
boundary fs p p
#-----------------------System Description from data file ------------------#
read_data final_data
#-----------------------Force Field Setting --------------------------------#
pair_style eam
pair_coeff * * Cu_u3.eam
timestep .0005 # 0.5 fs, as in metal unit time is in ps. 1ps = 1000fs
#----------------------To compute spatialy averaged temperature profile --------#
velocity all set {vel} 0.0 0.0 sum yes units box
fix 1 all nve
fix 2 all wall/reflect xlo {wallleft} xhi ${wallright} units box
compute ke all ke/atom
variable temp atom c_ke/(1.5*8.61e-5)
fix temp_profile all ave/spatial 25 8 200 x lower 3.0 v_temp file temp.profile units box
#---------------------To compute spatialy averaged stress tensor averaged diagonal elements--------#
compute peratom all stress/atom
variable meanpress atom -(c_peratom[1]+c_peratom[2]+c_peratom[3])/3
fix pressure_profile all ave/spatial 25 8 200 x lower 3.0 v_meanpress units box file pressure.profile
#----------------To compute x comp of velocity in spatialy averaged method--------------------#
compute vx all property/atom vx
fix velX_profile all ave/spatial 25 8 200 x lower 3.0 c_vx units box file velocityXcomp.profile
#----------------To compute spatialy averaged density------------------------------------------#
fix densityX_profile all ave/spatial 25 8 200 x lower 3 density/number density/mass file densityX.profile units box
fix densityY_profile all ave/spatial 25 8 200 y lower 3 density/number density/mass file densityY.profile units box
fix densityZ_profile all ave/spatial 25 8 200 z lower 3 density/number density/mass file densityZ.profile units box
#---------------To compute spatialy averaged atomic displacement-----------------------------------#
compute disp_per_atom all displace/atom
variable dis atom c_disp_per_atom[4]
fix dis_profile all ave/spatial 25 8 200 x lower 3.0 v_dis file dis.profile units box
#----------------To compute time average of global pressure ----------------------------------------#
variable mypress equal press
fix mypressure all ave/time 25 8 200 v_mypress file global_pressure.profile
#----------------To compute time average of global temperature --------------------------------------#
variable mytemp equal temp
fix mytemperature_profile all ave/time 25 8 200 v_mytemp file global_temperature.profile
#----------------To compute time average of kinetic energy ------------------------------------------#
variable myke equal ke
fix myke_profile all ave/time 25 8 200 v_myke file global_mykineng.profile
#---------------- To compute time average volume -----------------------------------------------------#
variable myvol equal vol
fix myvol_profile all ave/time 25 8 200 v_myvol file global_volume.profile
#---------------- To dump the trajectory -------------------------------------------------------------#
dump 1 all custom 1000 *.cfg id type xs ys zs id vx vy vz fx fy fz
dump_modify 1 element Cu
#------------------ thermo output ----------------------------------------------------------------------#
thermo 1000
thermo_style custom step temp pe ke etotal press vol lx xlo xhi
thermo_modify norm yes
#-------------------- Setting up of shock technicque-----------------------------------------------------#
restart 5000 *.restart
run 20000
write_restart restart.final
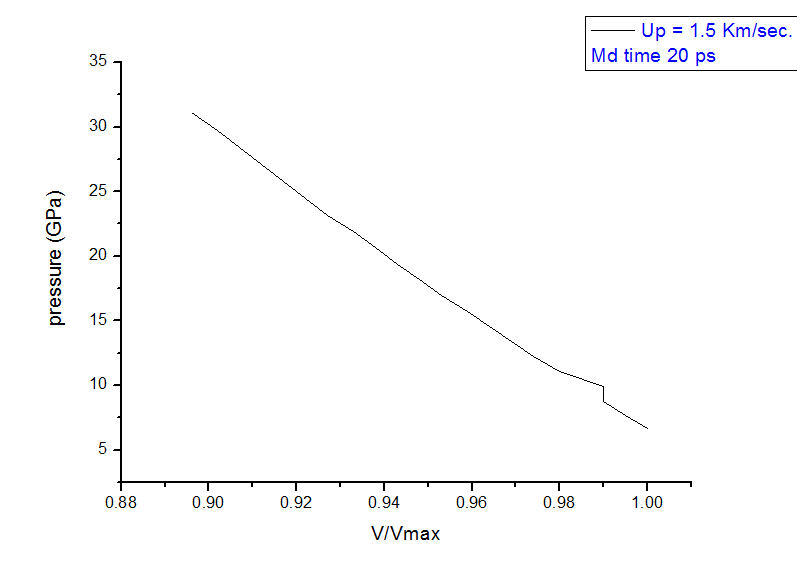
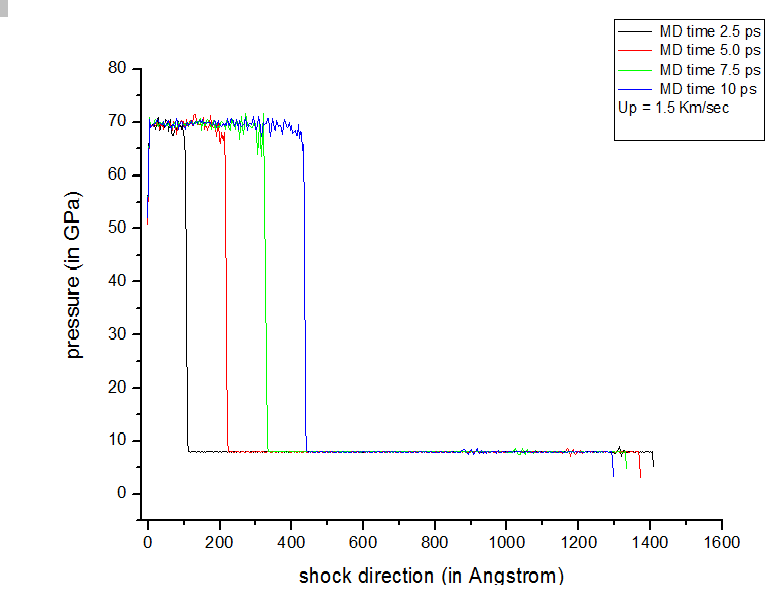
here I am setting the velocity along -X direction so, surely shock should propagate towards +X. So my plots showing that not BEHIND the shock, temp is increasing and pressure is decreasing, in-front the shock front. Behind the shock front temperature is decreasing and pressure is increasing, which is quit wrong. There is something which I am missing, please correct me.
I am attaching more plots to clear the fact more clear.
thanks