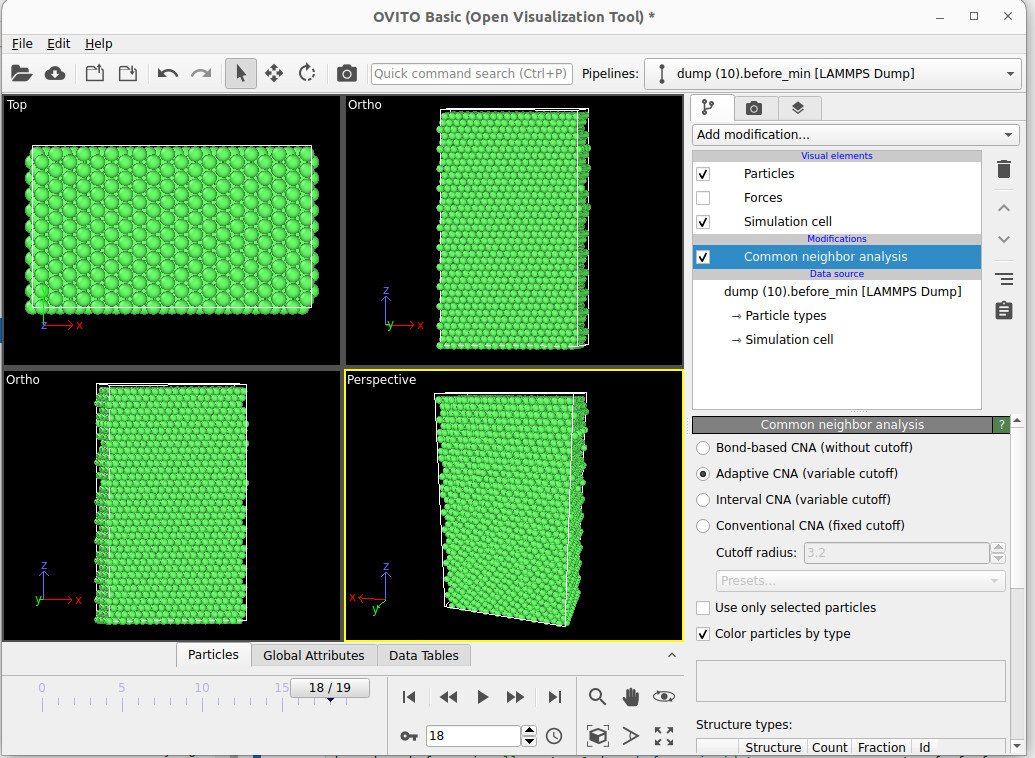
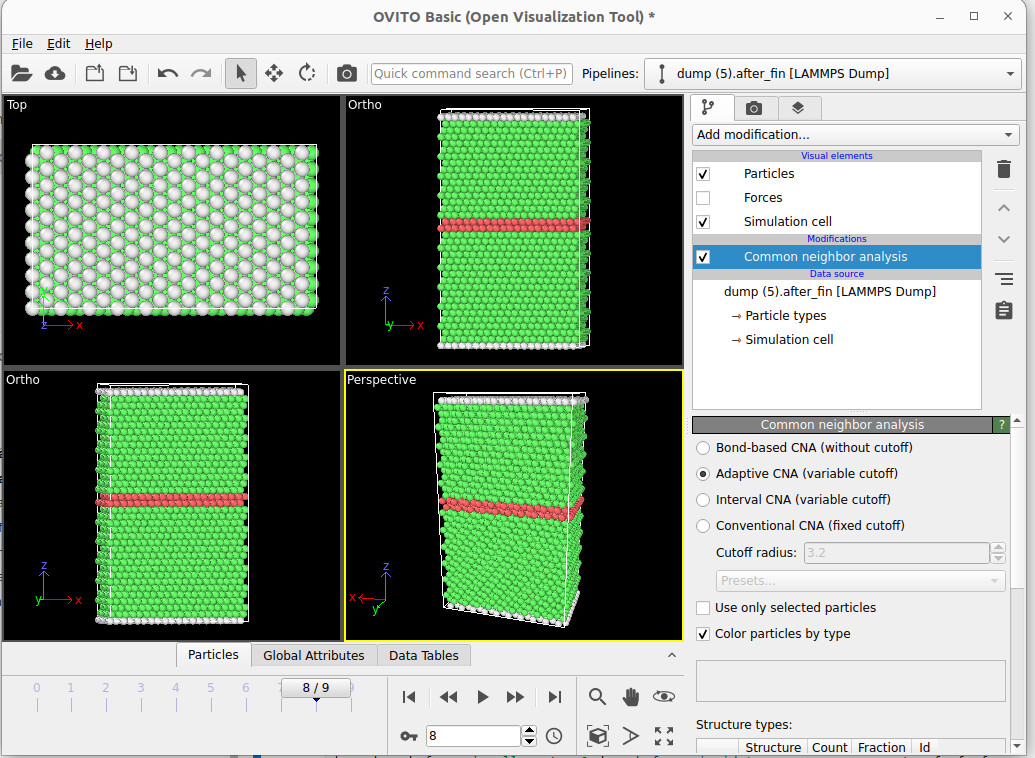
Hello,
I’m trying to create a stacking fault in a periodic Ni, first the lattice is perfect as attached, then after displacement, I was expecting a periodic stacking fault creation like in top and bottom but instead I find grey mismatched atoms, how to see the periodicity of the stacking fault? I also tried increasing the z dim of the simulation box but I couldn’t visualize it as attached. Thank you so much.
%%writefile NiNi.in
# --------------------- INITIALIZAITION ---------------------
clear
units metal
dimension 3
boundary p p p
atom_style atomic
# Create FCC lattice and define the simulation box
variable latparam1 equal 3.6
variable xdim equal ${latparam1}*sqrt(6)/2*10
variable ydim equal ${latparam1}*sqrt(2)/2*10
variable zdim equal ${latparam1}*sqrt(3)*12 # Increased from 6 to 12 layers
variable half_zdim equal ${zdim}/2 # New variable for half the z-dimension
region 1 block -.001 ${xdim} -.001 ${ydim} -.001 ${half_zdim} units box
region 2 block -.001 ${xdim} -.001 ${ydim} ${half_zdim} ${zdim} units box
region whole block 0 ${xdim} 0 ${ydim} 0 ${zdim} units box
lattice fcc ${latparam1} orient x 1 1 2 orient y -1 1 0 orient z -1 -1 1
# Define a straightforward displacement without modulus operation
variable x_displace equal -1*(${latparam1}/sqrt(6))
create_box 2 whole
lattice fcc ${latparam1} orient x 1 1 2 orient y -1 1 0 orient z -1 -1 1
create_atoms 1 region 1
lattice fcc ${latparam1} orient x 1 1 2 orient y -1 1 0 orient z -1 -1 1
create_atoms 2 region 2
# --------------------- EAM POTENTIAL ---------------------
pair_style eam/alloy
pair_coeff * * FeCrNi_d.eam.alloy Ni Ni
# --------------------- SETTINGS ---------------------
compute peratom all pe/atom
compute eatoms all reduce sum c_peratom
thermo 1
thermo_style custom step pe c_eatoms
dump dump_before_min all custom 1 dump.before_min id type xs ys zs c_peratom fx fy fz
run 1
undump dump_before_min
# Run initial minimization
minimize 1.0e-4 1.0e-6 100 1000
# Dump the configuration after minimization before displacement
dump dump_after_min all custom 1 dump.after_min id type xs ys zs c_peratom fx fy fz
run 1
undump dump_after_min
variable E equal "c_eatoms"
variable Eo equal $E
#variable E equal "c_eatoms" computes the initial energy of the model before any sliding is done
#E is necessary to store the initial energy in Eo
group bot region 1
group top region 2
displace_atoms top move ${x_displace} 0.0 0.0 units box
# Dump the configuration after minimization before displacement
dump dump_after_dis all custom 1 dump.after_dis id type xs ys zs c_peratom fx fy fz
run 1
undump dump_after_dis
fix 1 all setforce 0 0 NULL
min_style cg
minimize 1e-10 1e-10 1000 1000
# Dump the configuration after minimization before displacement
dump dump_after_fin all custom 1 dump.after_fin id type xs ys zs c_peratom fx fy fz
run 1
undump dump_after_fin
variable Ef equal "c_eatoms"
variable Cf equal 1.60217657e-16
variable A equal (${xdim}*${ydim})*1e-20
variable SFE equal ((${Ef}-${Eo})*${Cf})/${A}
#variable Ef equal "c_eatoms" computes the final energy of the system after sliding is done
#variable A is the area of the Stacking fault plane
#variable Cf is the conversion factor of electro volts to millijoules
#variable SFE is the stacking-fault energy of the system
####################################
# SIMULATION DONE
print "All done"
print "Initial energy of atoms = ${Eo} eV"
print "Final energy of atoms = ${Ef} eV"
print "Stacking-fault energy = ${SFE} mJ/m^2"