Hi all! This is my first post on matsci.org, so my apologies if I’m missing anything.
I recently set up Atomate, and I’m running a bandstructure workflow with Si to test my configuration. Here’s the script I’m using to add the workflow:
from pymatgen.core import Structure
from fireworks import LaunchPad
from atomate.vasp.workflows.presets.core import wf_bandstructure
struct = Structure.from_file(*path to Si POSCAR*)
wf = wf_bandstructure(struct)
lpad = LaunchPad.auto_load()
lpad.add_wf(wf)
This seems to run without error. Lattice parameters are reasonable, energy looks well converged, though I am getting higher stresses in the nscf line calculation (50 kbar diagonal, 10 kbar off-diagonal).
The band gap roughly matches the Materials Project value (0.62 vs. 0.61 eV), but my band structure does not resemble the Si bandstructure (mine has direct band gap, X point very different shape).
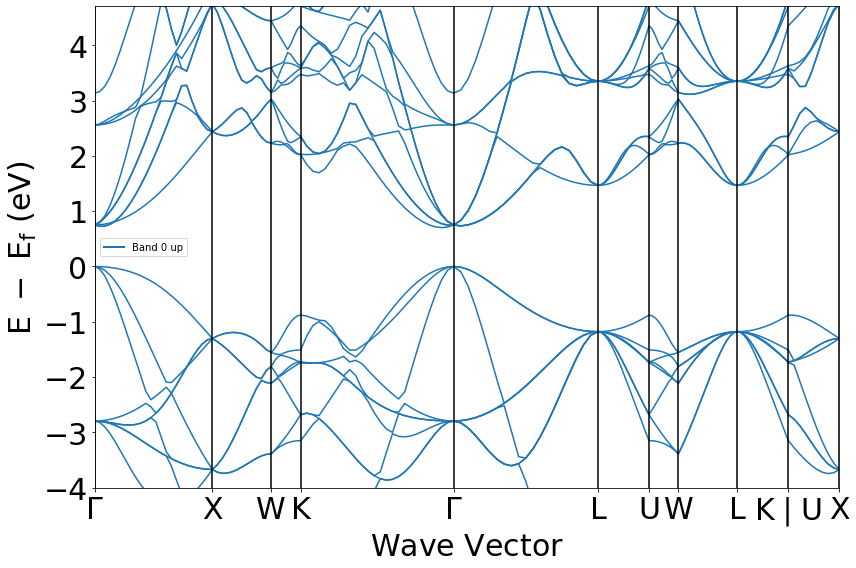
I’m plotting the bandstructure using this script, provided in the atomate tutorial:
from atomate.vasp.database import VaspCalcDb
from pymatgen.electronic_structure.plotter import DosPlotter, BSPlotter
atomate_db = VaspCalcDb.from_db_file(*PATH_TO_MY_DB_JSON*)
line_bs_entry = atomate_db.collection.find_one({'task_id':17})
bandstructure = atomate_db.get_band_structure(line_bs_entry['task_id'])
bs_plotter = BSPlotter(bandstructure)
bs_plotter.get_plot()
Any ideas why this might be happening? Thank you :]
