I’m having two problems with the Si DOS in MP (https://www.materialsproject.org/materials/mp-149/)
- The “interactive plot” just gives me the spinning molecule of death (technical issue)
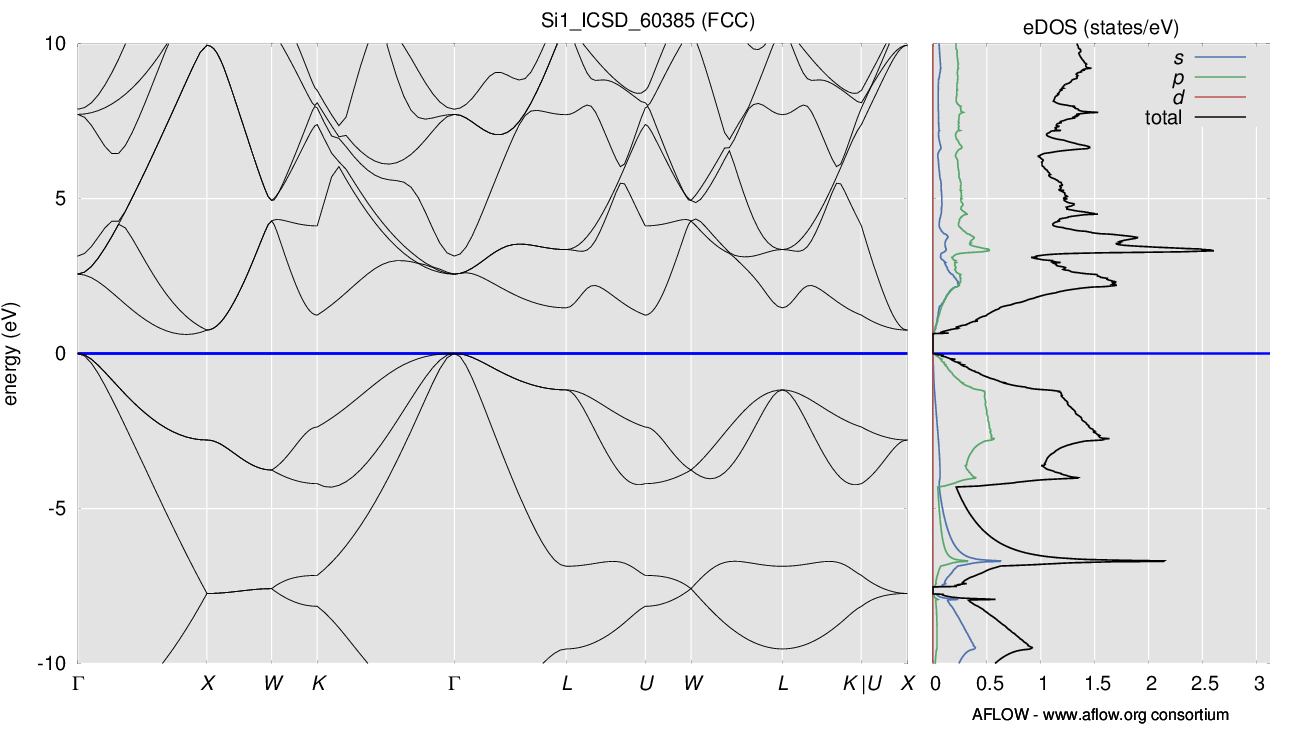
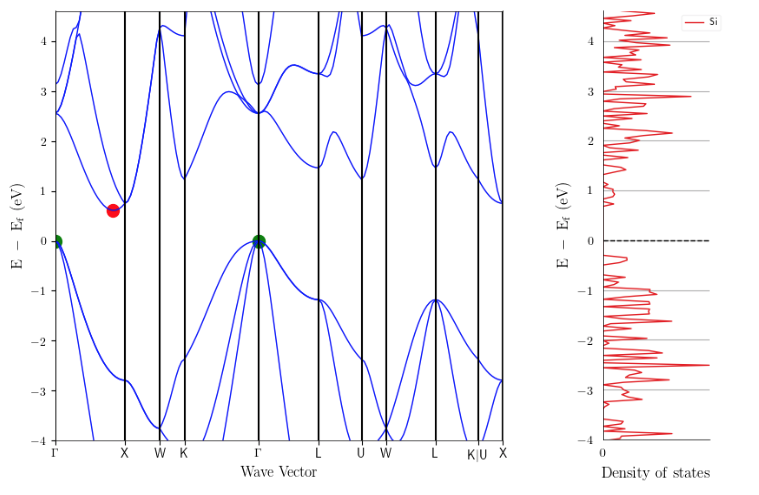
- The DOS itself, e.g., the valence band, just looks strange with all the various jagged curves. Sometimes the DOS drops down to zero (e.g., in the valence band between -0.5 and -0.75 eV) even though the band structure looks smooth. The DOS on AFLOWlib looks much smoother (http://aflowlib.org/material.php?id=aflow:a17a2da2f3d3953a) and more “correct”. (scientific issue)
Any idea what is happening?
AFLOW link doesn’t work for me.
Regarding 2., agreed the DOS looks terrible; this is not a Si-specific issue. I’d like to hear other opinions on this, but my understanding is (a) this is in part because we use VASP rather than a more sophisticated option, in particular, we could get nicer DOS by simply increase the NEDOS parameter, but this does require an additional VASP run (albeit a quick one, but still easier to do for new calculations) (b) beyond VASP, we should really use a better tool for post-processing, OptaDOS is fantastic but doesn’t currently accept VASP input, I think BoltzTraP interpolation would give a very nice DOS though and works well with our existing infrastructure. I’m not sure why we aren’t doing this, it might simply be a time issue.
We are looking at re-starting a band structure workflows, so we could incorporate these improvements for new calculations then.
The weird thing is that it does not seem(?) to be some kind of smoothing issue or even getting some quantitative features incorrect. The qualitative DOS just seems completely nonsensical.
For the AFLOW link - give it a minute. It’s not as fast as MP  I also attached the screenshot below
I also attached the screenshot below
(most of the other DOS on MP look a little jaggedy, but at least somewhat in-line with the band structure. The Si one just looks completely off)
Here’s the MP DOS and AFLOW DOS compared for example. Band structures match, but DOS are completely different.
Regarding 1., this should be fixed now. Note that the interactive version is processed by pymatgen.electronic_structure.dos.Dos.get_smeared_densities with sigma=0.1 for Gaussian smearing about the fermi level.
To update for anyone else reading this thread, one conclusion we came to is just that our densities of state are under-converged for the quality that we’d like to see on MP, so we’re going to investigate re-running these calculations.
Maybe you want to compare the number of k-points actually used versus that in the current atomate settings? My guess is that some of the calcs, particularly older ones like Si, probably have a mismatch between older settings and newer ones.
Yes, we were just discussing this. The current atomate settings might be sufficient.
Hi Anubhav,
as @shyamd can confirm, everything is ready to run interpolation of uniform bands for getting a much better dos via boltztrap2.
This is actually an old story…