Hello everyone,
I have 2 questions to ask:
first, what does * mean when used in lammps scripts like shown in manual : pair_coeff * * 100.0 2.0
and what do these mean?; sigma_14 and epsilon_14 which are to be defined in pair coeffs for lj/charmm/coul/charmm pair styles. how are they different from sigma and epsilon?
thank you.
The doc page for pair coeff and the lj/charmm potential
explain all of this. The * is a wildcard for many atom types.
The 14 are for 1-4 interactions between dihedral end atoms.
Steve
Hello everyone,
I have 2 questions to ask:
first, what does * mean when used in lammps scripts like shown in manual :
pair_coeff * * 100.0 2.0
and what do these mean?; sigma_14 and epsilon_14 which are to be defined in
pair coeffs for lj/charmm/coul/charmm pair styles. how are they different
from sigma and epsilon?
have you tried reading the documentation?
that is what it is for.
axel.
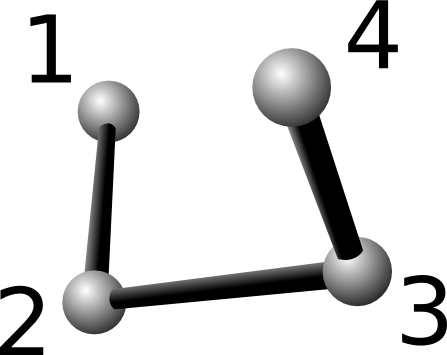
Conventionally, the "1-4" refers to the first and fourth atom in a
chain of consecutively bonded atoms (see attached picture). More
specifically, it refers to the first and fourth atoms which
participating in a dihedral interaction.
Typically users want to turn off or weaken the pairwise interactions
between these atoms, either with the "special_bonds" command, or by
using custom epsilon14,sigma14 parameters, (or perhaps by using custom
"weight factor" settings in dihedral "charmm", see below). Typically
1-2 and 1-3 interactions are also turned off (see
http://lammps.sandia.gov/doc/special_bonds.html). This is because
most users prefer to have the 1-2, 1-3, 1-4 interactions entirely
determined by the bond, angle, and dihedral forces (and not overly
influenced by the pair forces).
This means for every quartet of atoms in your "Dihedrals" section of
your data file, the LAMMPS the code which calculates the dihedral
angle forces, also (re)calculates pairwise non-bonded interactions
between the first and fourth atoms in this quartet using the optional
eps14 and sigma14 arguments you pass to the "pair_coeff" command.
(For this to work, you have to use the "charmm" dihedral style
{...incidentally usually I set the "weighting factor" to 0.0}) If you
leave out these parameters, the regular epsilon and sigma coefficients
are used (however the interaction is usually subsequently weakened or
disabled by your special_bonds or "weight factor "settings). It is a
little confusing because there are multiple similar ways of doing the
same thing. I hope this helps.

Thank you andrew
It was really helpful.
I did read the documentation but i didn’t understand it(specially about sigma and epsilon 14)
Reza
I did read the documentation but i didn't understand it(specially about
sigma and epsilon 14)
then you need to learn more about force fields
and particularly the CHARMM force field.
this is outside the scope of LAMMPS but *inside*
the scope of a text book on MD and molecular
simulations. users of all MD packages are expected
to educate themselves about the underlying theory
and models. the documentation contains some
recommendations and original references. a
quick web search will provide you with many,
many more suggestions.
axel.
Of course.
Thank you for your help. I will surely do it.
Reza