Hello all, I am trying to repeat a simulation in an article named ’ Investigation of PTFE Tribological Properties Using Molecular Dynamics Simulation’. When I built the model and simulate it with the same parameters, the outcome ‘Normal force’ and ‘Friction force’ have not the same tendency with the original simulation. My ‘Normal force’ is nearly straight and my ‘Friction force’ is close to zero. Instead, both forces in the article are stable growing.
How can I fix it?
The model shown as the following.
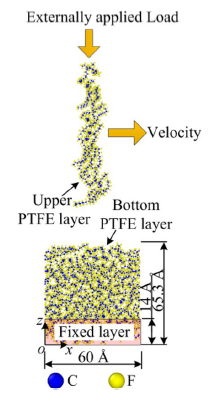
The material is ptfe. My model is as big as it. The external load is 1MPa and the x velocity is 0.147m/s. Here is my input:
boundary p p p
units real
dimension 3
atom_style full
pair_style lj/class2/coul/long 10.0
bond_style class2
special_bonds lj/coul 0.0 0.0 0.5 angle yes dihedral yes
angle_style class2
dihedral_style class2
improper_style class2
kspace_style pppm 1.0e-5
read_data layer.data
region downfix block INF INF INF INF INF 14 units box
region downt block INF INF INF INF 14 34 units box #downt
region downfree block INF INF INF INF 34 90 units box #downfree
group downfix region downfix
group downt region downt
group downfree region downfree
region up block INF INF INF INF 90 INF units box #up
group up region up
neighbor 3.0 bin
neigh_modify every 1 delay 0 check yes exclude group downfix downfix
variable FzC equal -0.02158
variable FzF equal ${FzC}*18.9984/12.0107
group C type 1
group upC intersect C up
group F type 2
group upF intersect F up
minimize 1.0e-4 1.0e-6 100 1000
timestep 1
thermo 100
compute myTfree downfree temp
compute myTt downt temp
compute myTup up temp
compute fx downfix reduce sum fx
compute fz downfix reduce sum fz
compute 1 up group/group down
fix ave all ave/time 10 5 100 c_1[1] c_1[3] c_fx c_fz file ptfetribology.txt
thermo_style custom step c_myTfree c_myTt c_myTup c_1 c_1[1] c_1[2] c_1[3] c_fx c_fz
thermo_modify flush yes
velocity downt create 300 5645354 temp myTt loop local dist gaussian rot yes
velocity downfree create 300 5654543 temp myTfree loop local dist gaussian rot yes
dump mydump1 all atom 1000 dump.friction.*
fix fxnvt1 downt nvt temp 300.0 300.0 100
fix fxnvt2 downfree nvt temp 300.0 300.0 100
run 100000
unfix fxnvt2
unfix fxnvt1
fix nvt_t downt nvt temp 300.0 300.0 100
fix nve_free downfree nve
fix 1 downfree langevin 300.0 300.0 100.0 48279
velocity up set 0 0 0 units box
fix setf up setforce 0.0 0.0 NULL
fix avef1 upC aveforce NULL NULL {FzC}
fix avef2 upF aveforce NULL NULL {FzF}
fix fxnve up nve
run 1500000 #1500ps
unfix setf
unfix avef1
unfix avef2
unfix fxnve
fix move up move linear 0.0000147 0 0 units box
run 500000 #500ps
unfix move
fix move2 up move linear 0 0 0.0001 units box
run 700000 #700ps
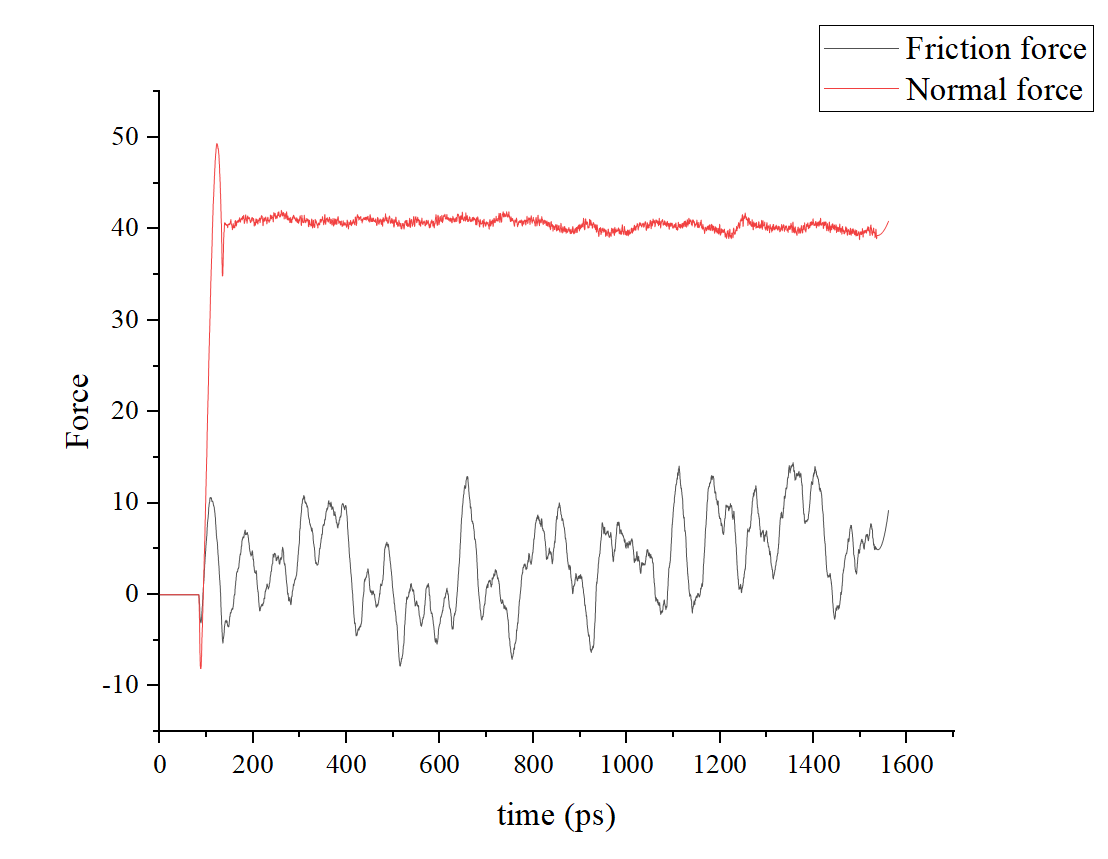
My ‘Normal force’ is calculated by c_1[3] and ‘Friction force’ is c_1[1] as shown in the following pictures.
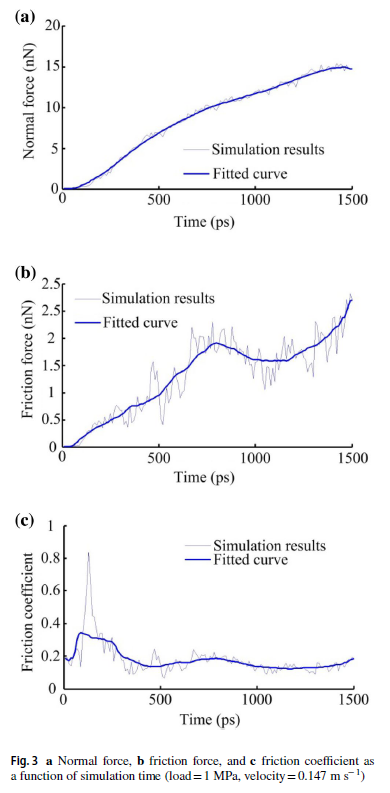
Here are the pictures in the article.
How can I fix my input to simulate the similiar results? I have been confused for several weeks. Thank you for any advice!