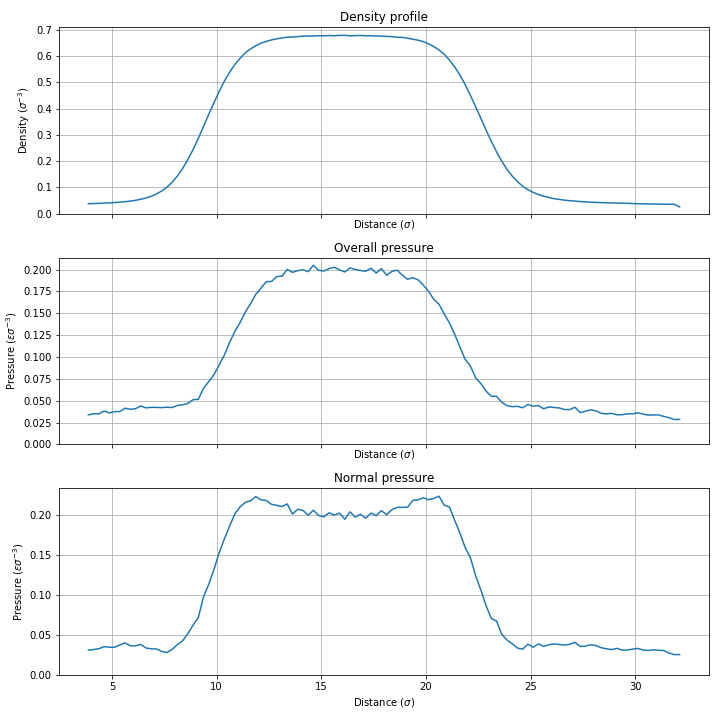
Hi all,
As part of a bigger project I'm trying to establish the saturation pressure of a Lennard-Jones fluid, or, more specifically, the truncated and shifted Lennard-Jones potential (implemented in LAMMPS as lj/cut). It is well known in literature that quantitative thermodynamic properties depend on the truncation point of the potential [1], so I thought the most straightforward method to establish this for my system is to run some NVT simulations myself for different densities while outputting the pressure.
For a vapour in equilibrium with its liquid phase, the pressure should be constant and equal to the equilibrium vapour pressure (saturation pressure) of the fluid. So as long as I'm in the coexistence region, the pressure of my system should be independent of the overall density.
I found this NIST data with accompanying LAMMPS input script (very convenient) that basically does this [2]. The only difference is that they use the linear force-shifted LJ potential (formerly implemented as lj/sf, nowadays it's lj/smooth/linear) instead of lj/cut.
Now the problem: I can't manage to produce the constant pressure behaviour that I expect. Depending on my boundary conditions and potentials, the pressure of my system either increases or decreases with overall density. Neither are physically correct behaviour for such a system, to my understanding at least.
My findings are as follows:
With lj/cut (cut at 2.5 sigma) as potential, PBCs in xyz, I find that the pressure *decreases* with increasing density. At an overall density of 0.185 I have a pressure of ~0.02. When I double the density to 0.37, the pressure reads as ~0.007.
With the cut-off at 5 sigma, I managed to produce *negative* pressures at one point. I don't understand how this is possible for a Lennard-Jones fluid.
If I use the lj/smooth/linear potential like the NIST people do, the differences in pressure are smaller, but still not constant like I would expect:
density pressure
0.1 0.050
0.185 0.047
0.37 0.040
If I make one dimension (z) fixed and install a wall at the top and bottom of the box and add a small gravity force (0.01) to get layering (I did this so I can easily analyse the densities of the two phases separately), get *increasing* pressure with density:
density pressure
0.1 0.041
0.2 0.046
0.3 0.059
All simulations are performed at a temperature T=0.85 (subcritical, of course) and all numbers are in reduced LJ units.
At this point I don't really know what to make of this. Are my assumptions correct? Is the problem in the pressure calculation? Is this an artifact of the (truncated) Lennard-Jones potentials? Something else even?
Ideas are appreciated.
Lars Veldscholte
[1] Smit, B. Phase Diagrams of Lennard‐Jones Fluids. The Journal of Chemical Physics 1992, 96 (11), 8639–8640. https://doi.org/10.1063/1.462271.