Hello everyone,
first off let me preface this query with the fact that I have experience with different atomistic simulation packages, but only just recently started to learn to use the LAMMPS package.
The version I am using is the stable version (9 Dec 2014), which I run on a linux based cluster of our university (should any further details be required with regard to the system specifications let me know). Most test calculations have been performed on a single core for small systems.
I am interested in simulating free surfaces for SrTiO3 using pair potentials, to this end I have been successful in simulating 3D periodic bulk SrTiO3 with the potential of choice using the Ewald long-range solver for the Coulombic interactions and a Morse potential for the short-range interactions. I use the HFTN minimizer as it yields the lowest resulting cell pressure (in comparison to the cg solver). I assume this is simply a result of how the solvers calculate the pressure.
Due to personal preference I use the fixedpoint 0.0 0.0 0.0 command in the box/relax fix. Once I have optimized the bulk structure, I replicate the cell to the desired size (for the test caluclations I use “replicate 4 4 4” which is a cell with 320 atoms). As I use the Ewald summation for the Coulomb interactions, I implement the “kspace_modify slab 3.0” option and change the boundary condition to “boundary p p f” and accept that I am restrained to orthogonal boxes as SrTiO3 has a cubic structure. Is there any information with regards to when triclinic boxes within the slab method will be implemented in the LAMMPS code?
The problem I encounter is that in some cases after the optimization is completed I get this error message:
“ERROR: Atom count is inconsistent, cannot write data file (…/write_data.cpp:149)”
I then reran the simulation with the added command “thermo_modify lost ignore” and view the resulting structure from the dump file and find that the optimized structure appears reasonable however a few atoms were relaxed to a position outside of the z-bounds of the box. If I understand the slab option correctly, then it implements a vacuum in the non-periodic dimension which is reproduced periodically due to how the slab method works. Hence, the loss of atoms confuses me. Especially since the error is not always present/consistent, as it most likely depends on the initial configuration (whether the atoms are located very close to the box bounds or not).
In one case the error appears if I refresh the neighbor list frequently (neigh_modify every 1 delay 0) and does not appear if I leave the neighbor settings at their default values. However, I have not found a way to reproduce the error in a consistent way.
Is there away to ensure that the error does not occur? Is it safe to assume that if the error does not appear that the simulation was successful?
I also tried to us the box/relax fix combined with the slab option to relax the periodic directions (x and y) with the following command:
“fix box_relax all box/relax x 0.0 y 0.0 couple xy vmax 0.10 nreset 1 fixedpoint 0.0 0.0 0.0”
Although the simulation completes, it does not change anything not even the atom positions as with the run without the box/relax fix. I do not know what is happening here, any help/explanations would be appreciated.
While reading the documentation of the slab command I came across the possibility of implementing a wall fix in order to prevent particle migration beyond the initial z-bounds. From my limited understanding of wall fixes, I believe a rough wall could fulfill the requirement of not losing atoms, but I am unsure whether it will allow the relaxation of these atoms as a free surface. Any additional information on this matter would be of great help to me.
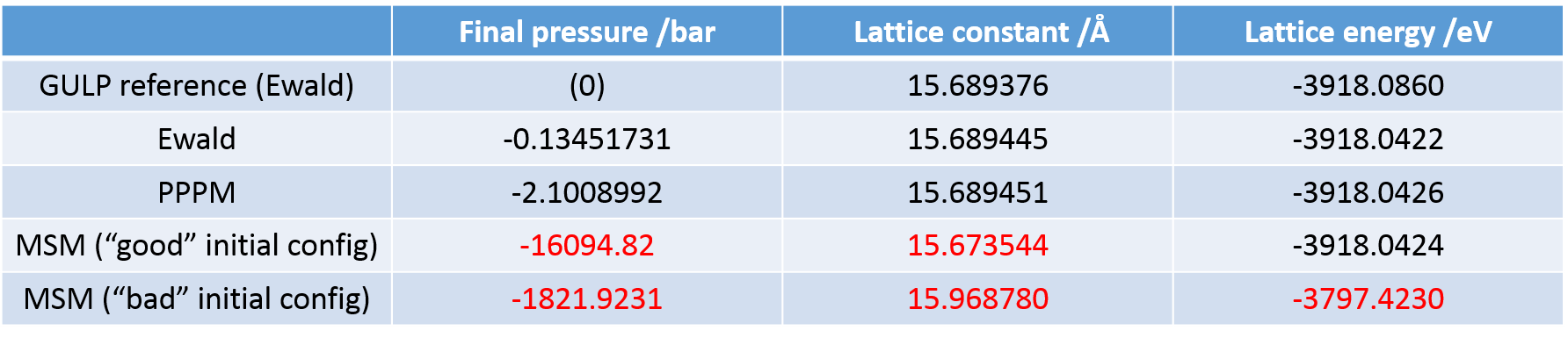
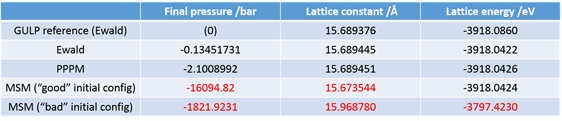
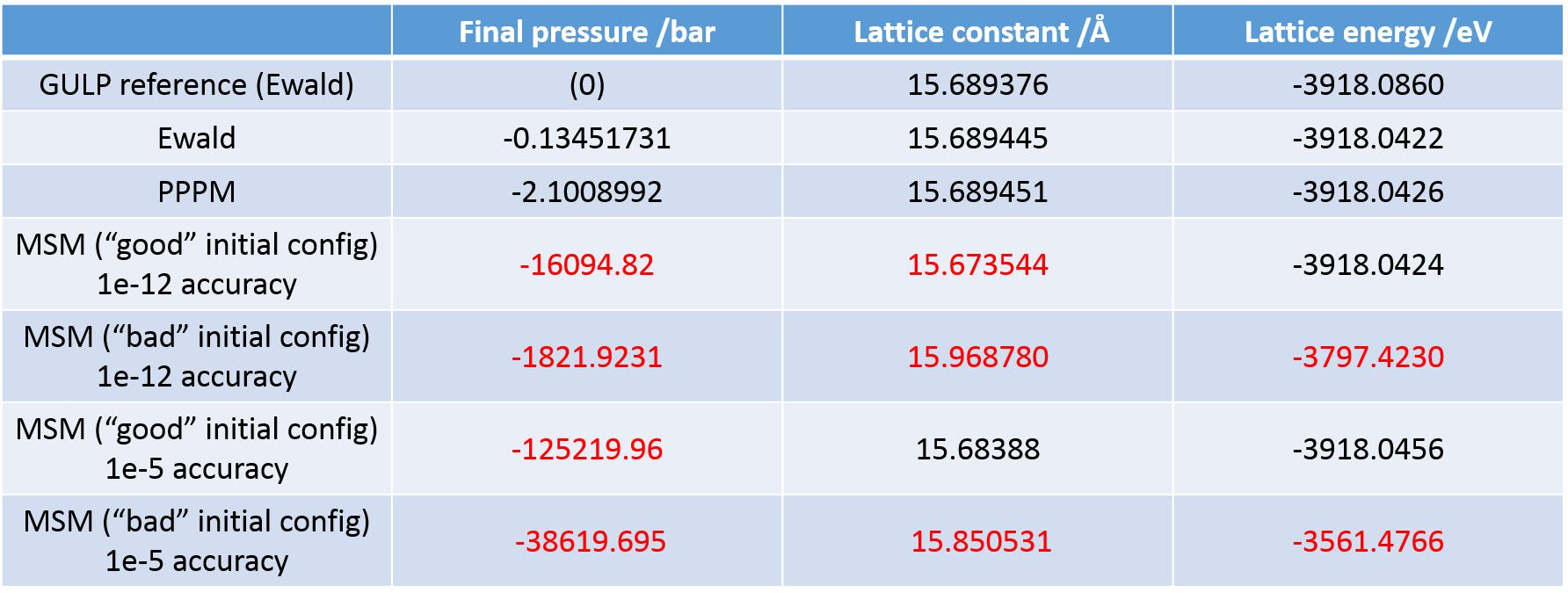
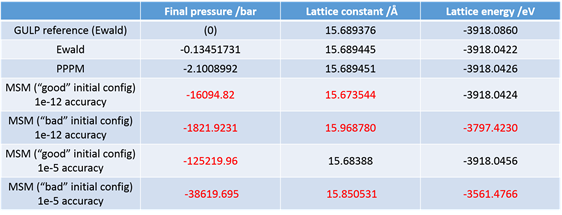
On a different note, I tried to implement the MSM solver for the long-range interactions for the same system as it allows non-periodicity in all three dimensions for triclinic systems, and encountered some difficulties in obtaining the same energy and box size I would get using the Ewald solver for the periodic bulk cell with the same initial data input. Furthermore, the final values given by the solver differ significantly depending on the initial configuration I input for the calculation (in this case I isotropically varied the initial box size). This inconsistency would prohibit the use of the MSM solver for the investigations I intend to perform. I use an accuracy factor—although it appears to be computationally expensive—of 1E-12 (“kspace_style msm 1.0e-12.0”). Is there any known reason for such discrepancies?
Even when I use a starting configuration close to the minimum, I do not obtain the value I would expect from using the standard Ewald summation.
Finally I had a question with regards to visualizing the slab simulations in Ovito (v2.4.2). As I understood it from the manual the “kspace_style slab 3.0” command internally inserts a vacuum in the simulation cell in order to obtain the free surface, however when I load the dump file into Ovito and show “periodic” images the inserted vacuum is not present (as it is not included in the dump file). Is it possible to dump the slab geometry with the vacuum portions in such a manner that it can be visualized with typical viewing programs?
For example input files see attachments:
*.lat is the input lattice via read_data.
*.in is the input script as was used in my test calculations
sto_bulk.lat was also used with sto_bulk_msm.in.
So to summarize my questions:
-
Is there any news on when triclinic box support will be implemented for the “kspace_modify slab 3.0” option?
-
Is it possible to reduce the possibility of atom loss due to the initial positions being close to the cell edge? Is it safe to assume that if the error does not appear that the simulation was successful?
-
Can an implementation of the wall fix be helpful in this case, and will it be equivalent to minimizing a free surface? If so, what kind of wall fix would be ideal?
-
The anisotropic box/relax fix does not seem to work with the slab option.
-
What is the reason for the different results obtained when using the MSM solver? Can it be corrected/fixed?
-
How can one best visualize the slab configuration when simulating surfaces?
Thank you in advance for your help. Should there be any required information missing, I will be happy to supply it.
Sincerely,
Amr Ramadan
sto_bulk_msm.in (1021 Bytes)
sto_surf.in (1023 Bytes)
sto_surf.lat (55.7 KB)
sto_bulk.in (984 Bytes)
sto_bulk.lat (1.16 KB)