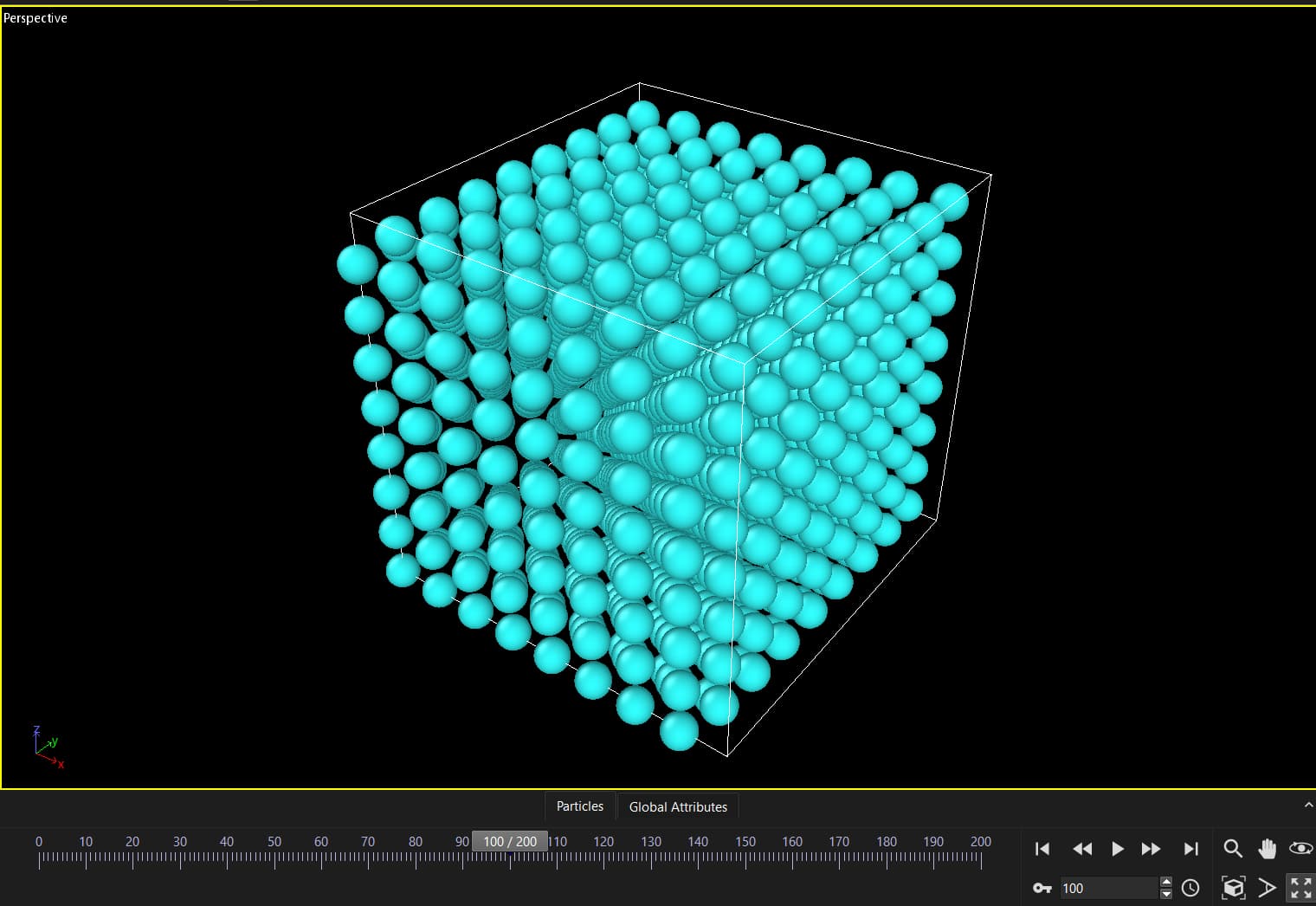
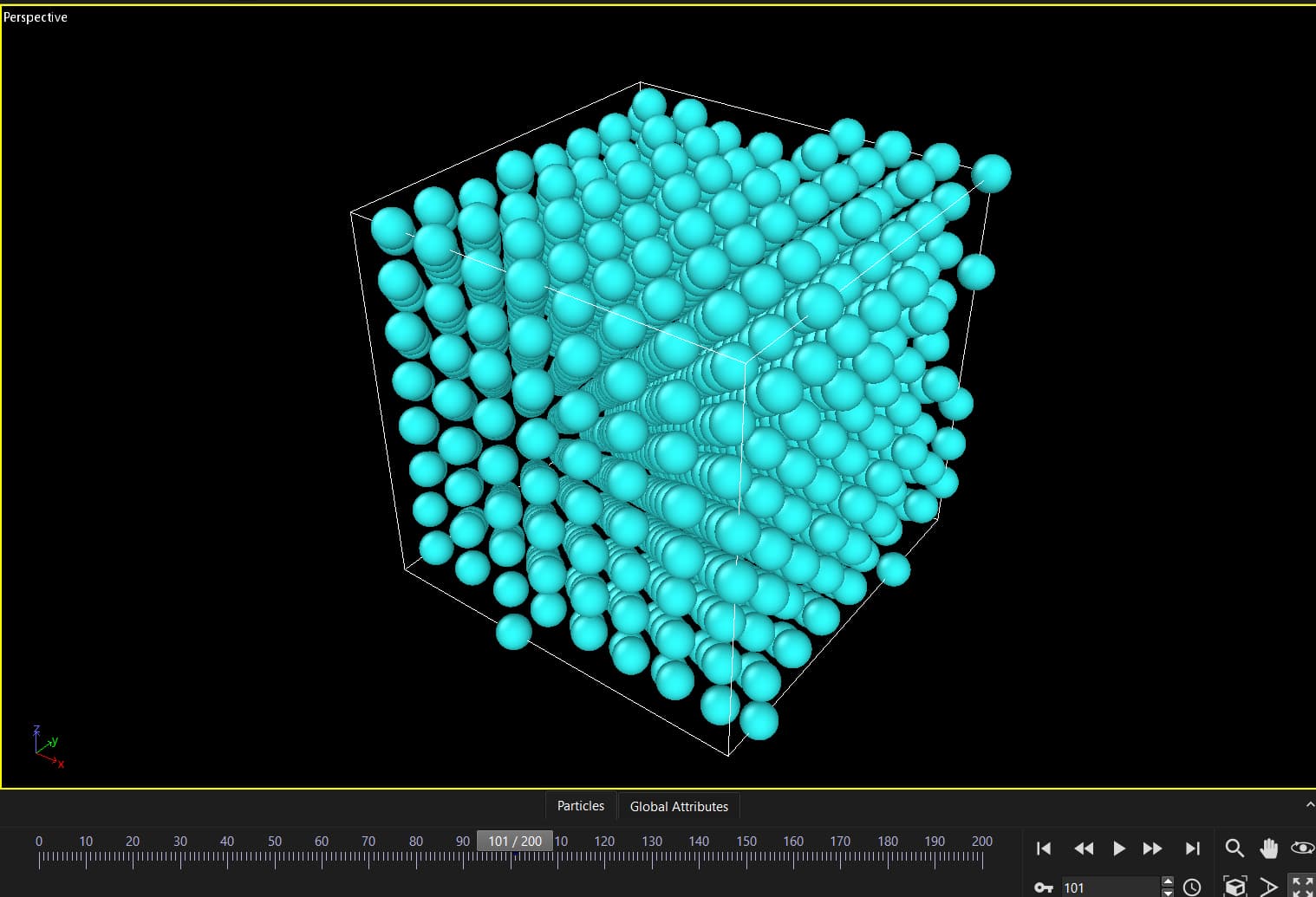
It turns out that my simulation box with temperature gets deformed between two runs with nothing in the middle. This is the input:
units metal
atom_style atomic
boundary p p p
atom_modify map yes
lattice bcc 2.8664999
region R1 block 0 8 0 8 0 8
#region R2 block 2 6 2 6 2 6
create_box 1 R1
create_atoms 1 box
group Region1 region R1
#group Region2 region R2
#group thermoboundary subtract Region1 Region2
fix integrate all nve
fix reset all dt/reset 10 1e-05 0.001 0.028664999 units box
pair_style eam/alloy
pair_coeff * * fe_mendelev_eam_alloy.potential Fe
thermo_style custom cpu cpuremain step dt time temp etotal ke pe
thermo 1
dump simulation all custom 1 simulation.xyz id element x y z
dump_modify simulation element 26
velocity all create 200.0 1 loop geom
run 100
run 100
If you remove the line velocity all create 200.0 1 loop geom, this deformation disappears. However, this script contains the minimum lines to reproduce the issue and is part of a bigger one. I need to create a box at 100 K and thermalise it (first run) and then do other things with that box and run again (second run). I have to use the NVE ensemble.
Some of your atoms are very close to the box periodic boundaries, so tiny fluctuations are enough to move the atoms from one side of the box to the other. I see nothing wrong here, this not a “deformation”, just a visual effect.
I’m expecting some movement due to temperature, the issue is that it’s not smooth. From timesteps 1-100 atoms move slow, from 100-101 there is a big change in the positions. I don’t think this can be attributed to temperature because it’s not smooth, but rather something related to two consecutive runs.
Your thinking is wrong. The situation is as @simongravelle describes it. If this is more visible at the beginning of a run, then this is because that usually enforces a neighbor list rebuild and that will update atoms according to the periodic boundary conditions, which may mean to move them across the box and change their image flags. This happens particularly often when you are using an unshifted lattice and box boundaries on multiples of the lattice spacing, since then a lot of atom positions are exactly on the box boundaries and already the tiniest motion can move them out of the box. For efficiency reasons, LAMMPS does not enforce this, but atoms are allowed to move outside the box for a little bit until the next reneighboring.
You can enforce dump output remapped exactly to PBCs with dump_modify pbc yes (at extra communication and compute effort).