in LAMMPS by using LAMMPS commands, I only want to generate different concentrations for the upper and lower regions while taking wall atoms solid and constant, I tried to create it by the following commands, but it didn’t work, can you please guide me?
######### Simulation box ############
region box block 0 20 0 20 -0.1 0.1
region lower block INF INF 0 10 INF INF
region upper block INF INF 10 20 INF INF
region wall block INF INF 9 11 INF INF
create_box 2 box
create_atoms 1 random 200 13487 upper overlap 0.4 maxtry 50
create_atoms 1 random 200 13487 lower overlap 0.4 maxtry 50
create_atoms 2 region wall
Firstly, thank you for your response. I couldn’t increase the number of atoms using that command to achieve an arbitrary concentration of fluid. The atoms of the wall are situated between them.
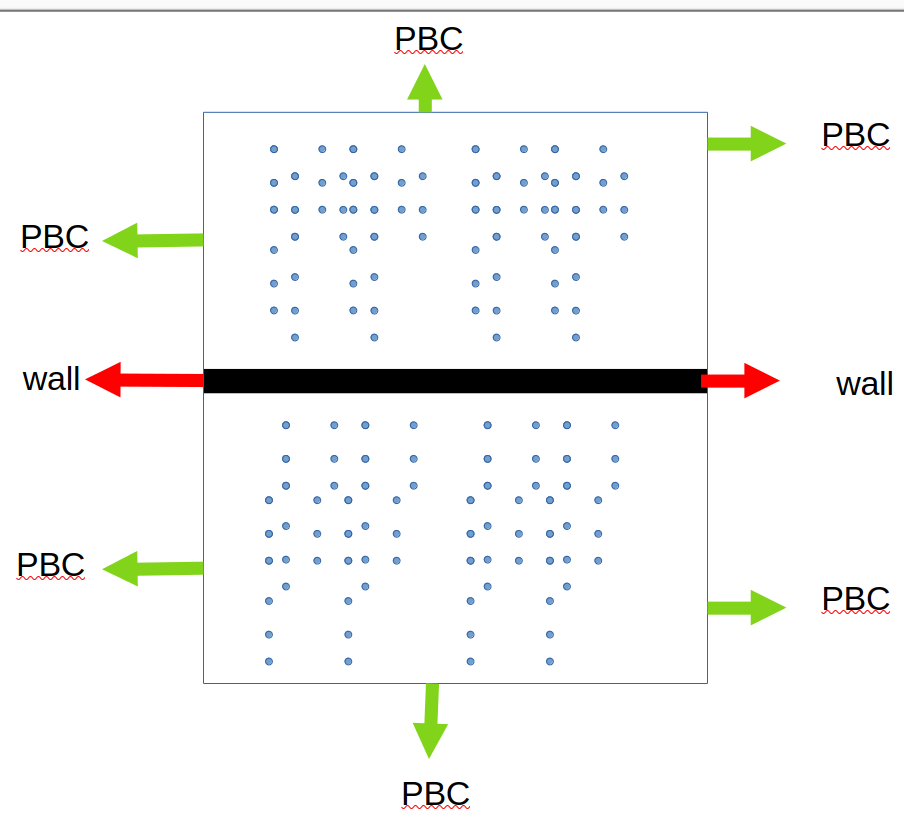
That wall separates the “upper” and “lower” sections only on one side, they are in free contact via the periodic boundary on the other side.
You can increase the number of atoms to arbitrary values, if you allow them to be in closer distance. That is just a necessity to get more atoms in a region (note you are using the overlap keyword). This is just basic math and obviously, you can easily get a density difference by simply creating fewer atoms. The term concentration doesn’t really make any sense here, since you seem to have only one type of atom on either side of the wall.
Bottom line, your problems are rather conceptual issues so you need guidance from your adviser, tutor or similar on the concepts of what you are trying to build. It doesn’t make sense to start simulations when you don’t yet understand such simple matters and it is not the job of the forum to train you in this.
Dear,
Firstly, I have a strong understanding of concentration as I have spent 15 years studying conceptual concepts of physics and math at high-ranked universities.
In this case, the volume is constant, and by increasing the number of atoms, I aim to increase the concentration of the fluid on both sides of the wall that separates them.
Whenever I increased the number of atoms, as follows:
region box block 0 20 0 20 -0.1 0.1
region lower block 0 20 0 8.99999 -0.1 0.1
region upper block 0 20 11.00001 20 -0.1 0.1
region wall block 0 20 9 11 -0.1 0.1
create_box 2 box
create_atoms 1 random 200 13487 upper overlap 0.8 maxtry 50
create_atoms 1 random 200 13487 lower overlap 0.8 maxtry 50
create_atoms 2 region wall
the following ERROR from LAMMPS pops up,
ERROR: Lost atoms: original 395 current 374 (src/thermo.cpp:494)
I thought my LAMMPS simulation box commands were wrong, so I attempted to modify them but encountered the same ERROR. It’s better to allow others to answer questions instead of answering them yourself, rather than making us feel foolish! Anyway, your behavior was reminiscent of an 18-year-old high school student.
Lost atoms means unphysically large forces have kicked some atoms an unphysically large distance. Usually this happens right after some atoms have become unphysically close.
This is information you would have found with a brief search of either the LAMMPS documentation or past forum posts. You have not provided enough information for any further useful feedback to be given.
Let’s get a few nomenclature and physics details right.
First, you have only one type of atom in those two regions. You cannot have a “concentration” since you have to have a solvent in order to get a “concentration”. When you have a fixed volume and put a varying number of particles into it, you are changing the “density”.
Second, with periodic boundaries and a single wall, there is no separation between the two sides of the wall. With periodic boundary conditions your origin is arbitrary and so is the location of the wall. Consequently, there is just one connected region on either side of the wall. From that
follows that you cannot have two different densities (or “concentrations” as you call them). At best there will be different initial values in the two areas.
@srtee already commented on this, so I don’t have to repeat that.
I fail to understand the logic behind this. You can easily check, if the command has done what you asked it to do by visualizing the resulting geometry. It seems you are mixing up correlation and causation. While changing the density can “trigger” lost atom errors, its cause are insufficient settings for such density, e.g. too large a timestep, too small masses, too steep a potential.
I am happy to comply with your request. I will configure my account so that the forum will not show me any of your messages in the future.
It is a pity that flame wars are against forum policy. Your post is providing plenty of ammunition for a rather juicy one that would also be entertaining to others.
When you complain about not being treated with the proper respect that you feel you deserve, you should refrain from exhibiting the same behavior yourself!