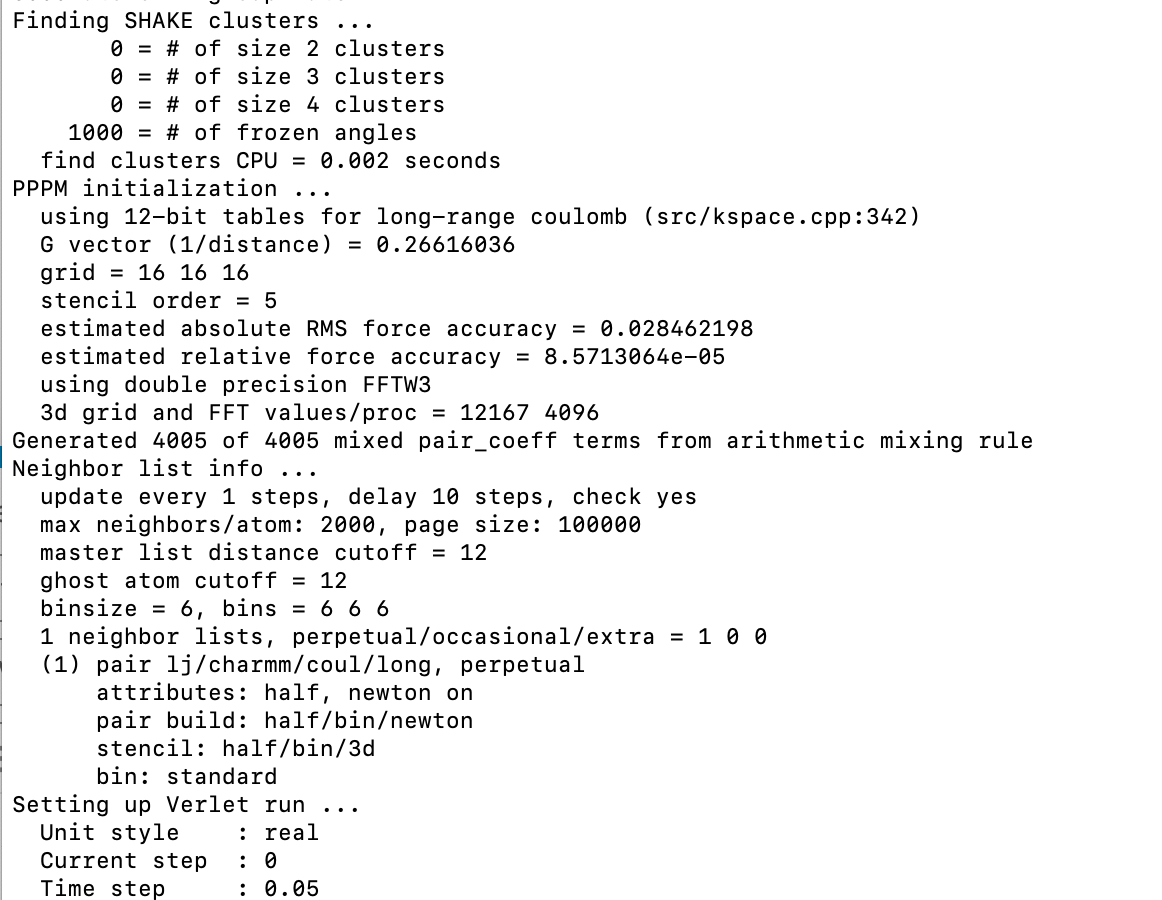
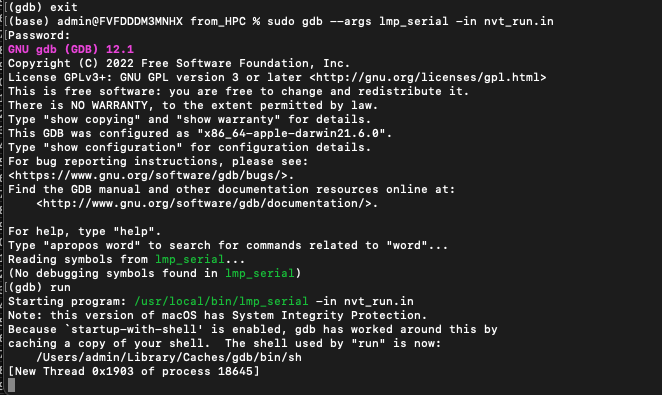
I tried running an NVT ensemble with the following command lines for a polymer-water-Sodium system and can’t seem to figure out why the system refuse to run. No error message was printed to enable me to decode the source of the issues. If you wish to rerun the simulation, please see attached the required files.
Thank you very much!!!
# ----------------- Init Section -----------------
include "system.in.init"
read_data "min.data"
include "system.in.settings"
# ----------------- Run Section -----------------
#
group water type 31 64
fix 1 water shake 0.0001 20 0 b 1 a 1
timestep 0.05
#
fix 2 all nvt temp 300.0 300.0 100.0
dump 2 all custom 1000 nvt_small_ts.lammpstrj id mol type x y z vx vy vz
dump_modify 2 sort id
thermo_style multi
thermo 10
restart 2000 restart1_polymer_1_nvt_2 restart2_polymer_1_nvt_2
#
run 400000
#
write_data nvt_small_ts.data
write_restart nvt_small_ts.restart
The output after executing lmp_serial < nvt_run.in
You should always report your LAMMPS version date and what hardware you’re running on. Looking through the forum, you’ll notice several posts with similar issues. On enterprise hardware (e.g., clusters), this is usually due to communication buffering. In some cases, you won’t see anything until the simulation completes. You can try running 1 or 10 steps instead of 400000 to test. That will also help test another option: if your hardware is local and fairly weak (I noticed you’re using lmp_serial), 400000 timesteps may take a long time, especially with long-range coulombic interactions. If you run a few steps, you can see how fast your system can run (e.g., in timesteps per CPU seconds).
Sometimes I’ve seen a segmentation fault kill LAMMPS without any error message, depending on the system. You can try running through a debugger, e.g. gdb -args ./lmp .... You can also try adding thermo_modify flush yes to your input, see thermo_modify command — LAMMPS documentation.
With a recent version of LAMMPS (15Sep2022 and later), you can use the -nonbuf (or -nb) command line flag to turn off all buffering for output to screen and logfile for testing/debugging of “lost” error messages.
This can happen if your system has overlapping atoms and otherwise extreme forces.
When using fix shake this can lead to problems with the Domain::minimum_image() function.
A simple correction for overlapping atoms would be to add the following line to your input:
delete_atoms overlap 0.2 all all bond yes
reset_atom_ids
This would remove all molecules causing overlap.
Another step you can do is run a minimization before running MD. With LAMMPS version 15Sep2022 and later, you can keep fix shake enabled, since there was feature added to replace the constraint with harmonic restraint forces; for older versions of LAMMPS you need to replace the force constants for the water bonds and angles (each type 1) with much larger values, so that the water geometry will be (mostly) preserved.
If the minimization runs find, but the code then gets stuck in MD, you can keep using the large force constants (w/o fix shake), but need to reduce the time step somewhat. At that point it is crucial to observe the potential energy, pressure and temperature. It should not deviate too far from the desired target. If it does or the potential energy and pressure are too extreme, then your starting geometry is likely flawed.
Thank you very much for the quick response. @Michael_Jacobs
My sincere apologies for not specifying the LAMMPS version. I am using LAMMPS (23 Jun 2022) version. Also, I have repeated the simulation for (timestep = 1 and run = 2) on an HPC with 1 node (64 CPUs) for 1hr and the situation is the same. The system looks like it hanged and I have attached the run and output files for your reference.
Using the same file as above, I added an extra line (thermo_modify flush yes) and saw no difference in the result. Also, I tried the debugger with gdb but I am not sure if I am doing it the right way. See the output below.
@akohlmey Thanks for the detailed explanation. I will try implementing your suggestions and update you if I experience any issues or success.