Greetings MATSCI Community Members,
As a newcomer to LAMMPS, I’m currently in the process of learning its intricacies. I have a few uncertainties concerning the outcomes of my MD simulations involving Dislocation Velocity. I would greatly appreciate any assistance or insights from you all in clarifying and comprehending these matters.
To aid in my learning, I’ve been utilizing the LAMMPS script provided on the Mississippi State ICME website (link: LAMMPS Dislocation Mobility - EVOCD).
In this script, I’ve created a periodic array of dislocations. The lower section of the simulation box remains fixed, while a uniform stress is applied to the upper side to induce dislocation movement. During the dislocation simulation, I’ve employed the Fix NVE method. I am doing simulation for Aluminium using the following potential : Zhou et al, Acta Mater, 49, 4005 (2001). The size of my simulation cell is 100X80X4.
For post-processing tasks, I’ve turned to OVITO and its Dislocation Analysis Filter. While visualizing the dump files in OVITO, a particular concern has emerged. The number of ‘other’ atoms, which contribute to dislocation behavior, seems to fluctuate across different time steps, as depicted in the images below:
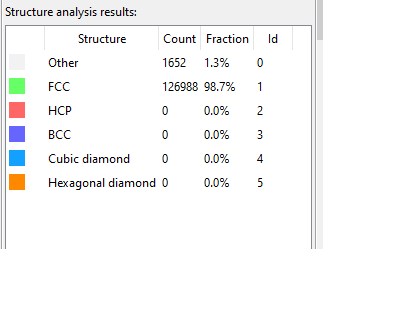
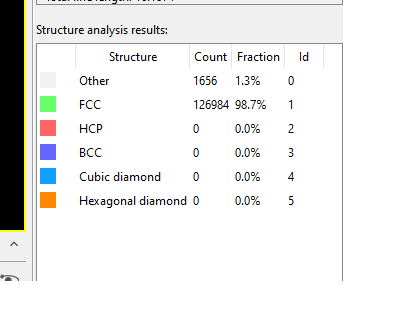
More often than not, these groups of atoms maintain a resemblance to the initial cluster. However, periodically, they transition into irregular structures as demonstrated below:

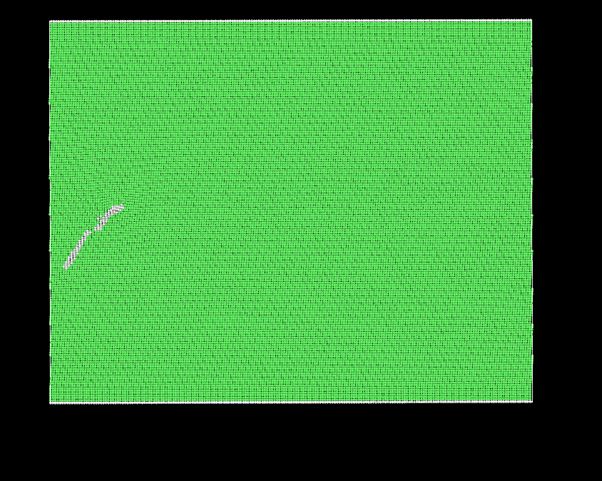


I’m uncertain whether such variations are anticipated within the LAMMPS framework or if there might be errors in my approach.
I’m reaching out to seek guidance on resolving this quandary. Your expertise and insights would be invaluable to me.
I extend my gratitude for investing your time and attention in this matter.
Best Regards,
Vikram Roy