Dear all, I am currently simulating the ionic liquid in the presence of an electrode in constant potential, however, I have to start from a very small time step like 0.001 fs, and after 5x10^6 steps, I was able to run with timestep 1fs, otherwise the atoms are going out of box. The force field parameters taken from lammps example graph-il file. The ionic liquid and electrode are as suggested in the example bmimpf6 and graphene. Kindly suggest some ways to get rid of this problem.
That is really not a question about LAMMPS but about your geometry and force field parameters. If both are correct and meaningful for your system, then you can run a compatible timestep. If you timestep has to be smaller then something is not quite right. There are multiple remedies starting from creating a better initial geometry, that have been discussed without end in this forum here. Please be so kind and look for it yourself instead of asking us to repeat the same stuff over, and over again.
It is not even about the force-field parameters – as I’ve checked myself quite a few times just this week, the ELECTRODE examples run just fine “out of the box” even on a laptop.
You may not have copied them correctly, or you may be attempting to apply them to a physically invalid configuration.
Dear Srtee, Thanks for your kind insights; as you have mentioned, the Lammps example works fine, but I could not understand how they have assigned the charge to the carbon atom, i.e., atom no 5. By what calculations did they determine the charge of carbon in data.graph-il file , all the carbon atoms don’t have the same charge. Kindly explain these to me. I am attaching my datafile here.
ele_edit.data (185.6 KB)
The carbon charges are not directly meaningful – these examples are meant for use with the ELECTRODE package, which dynamically reassigns charges to electrode particles based on an assigned potential difference, and thus the initial charges are not retained. (With the exception that if the conjugate gradient algorithm is used, a good choice of initial charges will speed up convergence on the first time step.)
Thank you for your explanation regarding the charge on Carbon atoms. I also could run the electrode package, but I could not obtain the expected rdf, i.e., the rdf should have some sharp peak near the positive electrode when taken concerning negatively charged ions. I have numbered the electrode as atom no 5, and the imidazolium ring as atom no 1. I am attaching the input file and graph here. I would be very grateful if you could explain the reason behind this.
in.conp (665 Bytes)
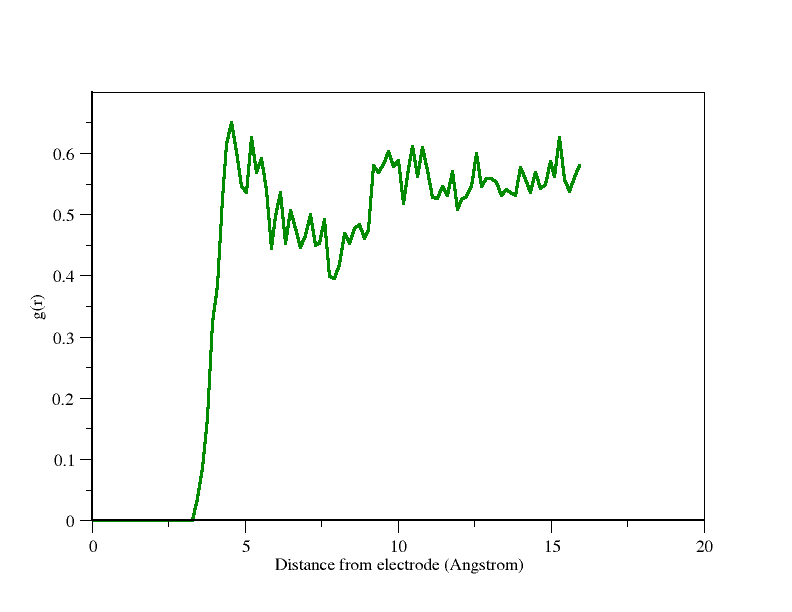
5000 steps is far too short to converge your results.
Please note that the examples folder primarily shows you what the correct syntax is – that is, what keywords to use with what commands to make a simulation run and not crash. They are not guides to running a scientifically valid simulation. If you are unfamiliar, you should start by trying to replicate a paper’s results, including the run lengths specified there.
There are three possible reasons:
- your force field parameters are not matching those of your reference or were not entered correctly
- your system is not yet equilibrated, i.e. the g(r) is changing
- your data acquisition is not sufficient, i.e. the g(r) plot is noisy
Thank you for your kind explanations, Akohlmey and Srtee. I would like to know, in the constant potential simulation, the charge on the top and bottom electrodes varies constantly. How do we extract the Capacitance from the output file, i.e., the log file?
As always, the capacitance is just the electrode charge divided by the potential difference. Your log files will tell you the average charge, and the potential difference is what you imposed.
Please note that, as usually happens in molecular simulations, the very small system size results in large fluctuations of observables. Please read and review the ELECTRODE literature to understand the theory behind this.
Dear akohlmey,
Thanks for the kind response. I have a doubt about equilibrating the system with electrodes. Following your advice, I tried to equilibrate it, but observed that I couldn’t apply NPT here, so what is the solution, i.e., a very long NVT? Again NVT is not enough according to procedure. Is there any tag in LAMMPS with which we can equilibrate the system with NPT?
we cannot perform NPT in p p f boundary conditions
You can, but you cannot couple to all three boundaries. See the fix npt documentation.
I am closing this topic now for two reasons:
- The discussion has gone far, far away from the initial discussion topic. In fact, it is hard to figure out which response your comments and questions are referring to (in part because you don’t quote the text, in part because you just keep piling on questions).
- It has become evident that you need proper in-person local tutoring. You are trying to use this forum as a substitute for an adviser or tutor and that is not its purpose. Nobody here has the time or cares enough about your research to spend that much time on your questions, and that specifically comes from level of experience not being on par with the research topic you have chosen.
So if you have more questions, please make sure that:
- you have researched the manual properly
- have a sufficient grasp of the theoretical background of your question
- provide sufficient details about your simulation with your question
- make sure your question is properly formatted and readable