Dear all,
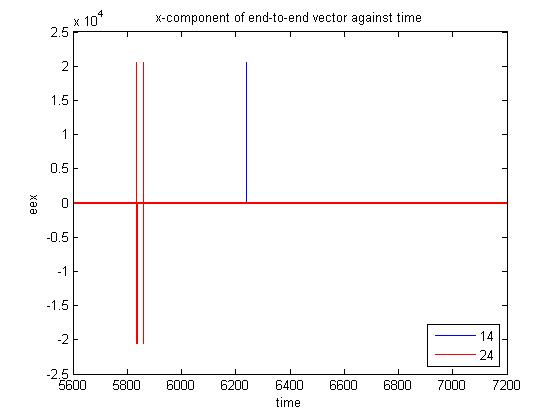
I wish to perform a Dissipative Particle Dynamics simulation of polymer chains migrating in an array of cavities in the presence of a steady external field. To do this, I am using a cubic simulation box of side length 20 with periodic boundary conditions in all 3 directions. There are 3 types of particles in the simulation. The solid mass comprises of 8818 particles arranged to form a spherical cavity. Also, there are 30 polymer chains with 10 beads each and 12481 solvent beads. Please find attached the LAMMPS input script (‘cavity.in) and data file (‘input_lam.dat’). When this simulation is run for more than 20 million time-steps with dt = 0.04, the component of the end-to-end vector of some chains along the direction of the field suddenly becomes very large (of the order of 10^4) at certain time-steps before returning to normal. I have attached a graph showing the variation in end-to-end vector of two chains (chains 14 and 24) with time. I am using chains with harmonic bonds. The force constant is 40 and the equilibrium bond length is 0.7. The problem does not arise in the absence of the external force.
I would appreciate if someone could clarify the following issues:
-
What could be the cause of this problem? I have checked the values of various thermodynamic parameters (temperature, pressure etc) and nothing seems amiss. Also, why didn’t the simulation stop with a ‘Bond/Angle atom missing on proc ‘ error message when the chains became abnormally large?
-
What can be done to rectify this problem?
Thanks.
Best,
Karthik.
cavity.in (784 Bytes)
input_lam.dat (1.06 MB)
Dear all,
I wish to perform a Dissipative Particle Dynamics simulation of polymer
chains migrating in an array of cavities in the presence of a steady
external field. To do this, I am using a cubic simulation box of side
length 20 with periodic boundary conditions in all 3 directions. There are
3 types of particles in the simulation. The solid mass comprises of 8818
particles arranged to form a spherical cavity. Also, there are 30 polymer
chains with 10 beads each and 12481 solvent beads. Please find attached
the LAMMPS input script (‘cavity.in) and data file (‘input_lam.dat’).
When this simulation is run for more than 20 million time-steps with dt =
0.04, the component of the end-to-end vector of some chains along the
direction of the field suddenly becomes very large (of the order of 10^4)
at certain time-steps before returning to normal. I have attached a graph
showing the variation in end-to-end vector of two chains (chains 14 and 24)
with time. I am using chains with harmonic bonds. The force constant is 40
and the equilibrium bond length is 0.7. The problem does not arise in the
absence of the external force.
I would appreciate if someone could clarify the following issues:
1. What could be the cause of this problem? I have checked the
values of various thermodynamic parameters (temperature, pressure etc) and
nothing seems amiss. Also, why didn’t the simulation stop with a
‘Bond/Angle atom missing on proc ‘ error message when the chains became
abnormally large?
your system will exhibit a drift in x-direction due to the added force
representing the external field. this will lead to your entire system
passing through perdiodic boundaries repeatedly. if you run for a very long
time this will overflow the image flag counters (which are limited to 10
bit signed integers).
you don't get a bond missing error, because LAMMPS will look for the
closest periodic neighbor, without consulting the image flags.
2. What can be done to rectify this problem?
you can add a restraining force to the part of the system that should
remain stationary, e.g. using fix spring with the tether option, if you
want to get rid of the drift.
if you prefer to keep the drift (remember, that the kinetic energy of the
drift is included in your temperature), you have to re-compile LAMMPS with
-DLAMMPS_BIGBIG so that image counters are stored in 21-bits signed
integers instead.
axel.