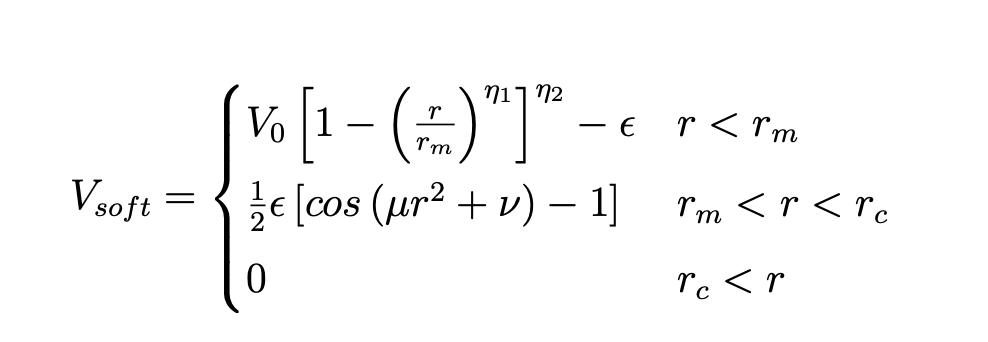Dear All,
I am simulating a polymer with 500 monomers which uses a custom written pair_style(soft_cut and soft_pot).These potential can be analogous to lj cut potential and lj attractive potential. So the model I am using for the Polymer is 2 types of particles say A and B. Type A monomer repel A and attract B. B interacts attracts B. Here Attractive is soft_pot and repel is soft_cut. I am also attaching the files that I have written.
So my simulation is changing the percentage of type B beads in the polymer and see how the scaling factors changes. For 10% of type B beads in a polymer of 500 particles the simulation for 10 runs and 10^7 steps and time-step of 0.05, wall time is around 3:03:17.
As I increase the percentage of B beads the simulations becomes slower to the extent that around 60% attractive it takes around 30 hours. There is an exponential rise in time. I understand that once there is an attractive potential there must more number of short range calculations to do which might increase some time. But 30 hours is somethings i cannot understand. I have attached a log file of 10th run of 40% type B beads, input file and the potential files that I have written, I also read that some potential are computationally expensive. I am adding a form of potential that I am using below. To get soft_cut part I just remove the part from r_m to r_c. Please tell me if I need to upload anything more. I also tried parallel processing, but since the number of atoms are so low per processor I tried to run this parallel but there was bottle necking. Any suggestions on what might be the problem are welcome!
Thank you for your time!! run_10.log (72.2 KB)
If you want to find out why your simulation is getting slower, you need to do some profiling to identify the part of the code that is taking more time.
A very coarse profiling information is given in the LAMMPS output at the end of a run. If you are running on a Linux machine, you can get more detail by using kernel-level profiling with “perf”, or you create an instrumented binary for profiling with “gprof”.
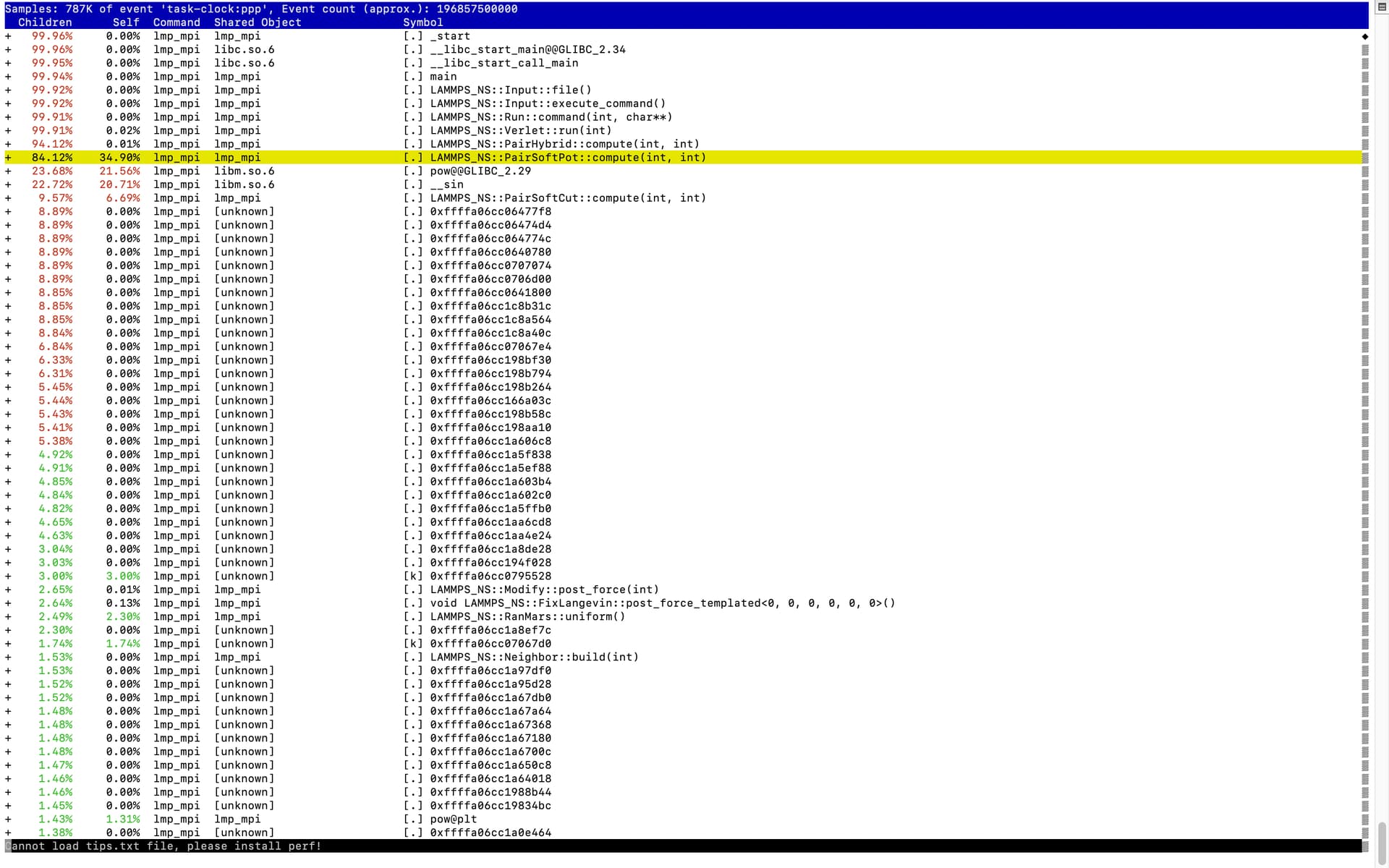
So I used perf to profile my code. I am uploading the screenshots of the perf report.
This screenshot is when there are 70% attractive beads. I see that PairSoftPot compute takes 34% of time in itself, where as for 10% attractive beads it was around 7% of time was spent in function itself
After looking in the code, to see the part which of the was taking more time I saw that square root operation was computationally expensive. There are two loops which take 20% and 14% respectively(as seen in screen shots) of the 34% of the time spent in SoftPotcompute. What I dont understand is why is square-root so computationally expensive and what I can do to make it faster.
I have added the screen shots of the functions which take a major part of calculation. Also instead of using input parameter from user I have directly used parameters in the code itself for force calculation as the potential parameters for this calculation are not going to change. This reduced 7% time, earlier it was 41% time spent in the code. Thank you for your time!
This is just a fact. Computing the square root is expensive. This is the reason why the commonly used Lennard-Jones potential is a 12-6 Lennard-Jones potential (9-6 would be more accurately representing the atom-atom interactions), because it can be programmed in such a way that it no square root is required. For the same reason, a Morse potential is not used (even though its shape is far closer to typical atom-atom interactions) because it requires the computation of exponential functions in addition which is even more expensive.
In your specific case, you are losing even more time from the use of the power function (since that corresponds to an exponential and a logarithm).
The only approach I know to speed this up is to avoid those computationally expensive operations. There are two possible ways: 1) try to reformulate your equations so that the terms using expensive function calls are precomputed (this is not always possible), 2) if the interaction is pair-wise additive, try using pair_write to generate tables for each pair of atom types and then use pair style table (which is usually more noisy and thus has worse energy conservation). Its cost and accuracy depends on the number of tabulation points.
Overall, the growing computational cost is due to the increasing number of neighbors resulting from more atoms with attractive interactions and there is nothing that you can do about this, since that is a consequence of the physics.
I would advise you to write a unified pair style instead of using pair hybrid. If you are later going to use different combinations of attractive, repulsive, etc – write those as unified pair styles as well.
I say this because if you have very expensive pair calculation functions, the last thing you want to do is trawl the neighbour list twice per time step and recalculate all of the pair distances you’ve already spent time calculating in the first pair style (unless I am misunderstanding pair hybrid).
The overhead for pair style hybrid is less than that since the neighbor list code will generate so-called “skip lists” for each hybrid sub-style so that the number of pairs is the same, only the effort of generating the skip lists is added.
This is different when you are combining, e.g., lj/cut and coul/long with hybrid/overlay to emulate pair style lj/cut/coul/long.
Well, I was in a mathy mood tonight, so I worked out how to get to the same end result with as few pows as possible – I got it down to two, while eliminating a square root.
Please note that you should understand the philosophy of reusing expensive calculations as much as possible. I don’t guarantee that I’ve got the math right, and it’s on you to double check that the calculation is correct and implement it correctly.
Can you tell how to write a custom pair_style with full read, write_restart, etc support?
Also, if there is any thread, where someone teaches how to create custom pair_style, bond_style, fix etc please share