Hi everyone,
I want to perform a simulation with a piston on fused silica. I have tried read the fused silica file, extend the dimensions of the simulation box and create atoms of the piston in the upper part of the box (blank region) but it gave an error. So I had to create a piston of Si atoms, take all the information of these atoms from the piston.lmp file and “merge” them with the atoms in the fused_silica.lmp file. So I have now this structure as an input for the simulation
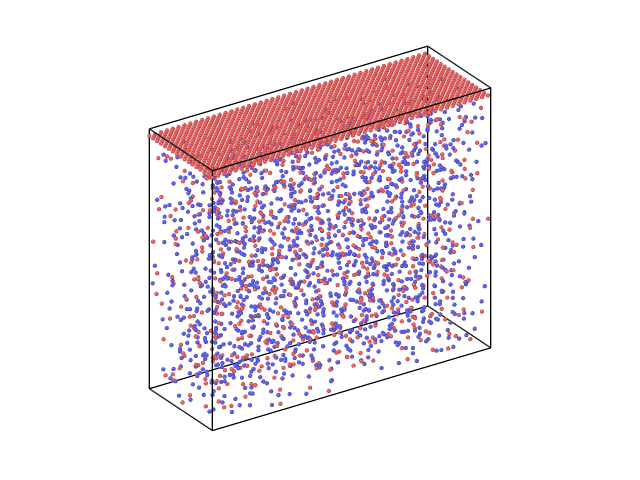
But as soon as I start the simulation I get this error:
ERROR: Non-numeric pressure - simulation unstable (src/fix_nh.cpp:1043)
Last command: run ${time_eq}
So I have 2 questions now:
- Is it possible to read a file with read_data command and then create some new atoms, or atoms cannot be created in a box already consisting of atoms ?
- What’s the reason of the error of the non-numeric pressure and how to get rid of it.
The input script is below:
clear
units metal
dimension 3
boundary p p p
atom_style atomic
#atom_modify map array
variable stemperature equal 300 # temperature in kelvin
variable alattice equal 3 # lattice constant (unit A)
variable myseed equal 12345 # the value seed for the velocity
variable xmin equal 0.0 # size in the x-direction
variable ymin equal 0.0 # size in the y-direction
variable zmin equal 0.0 # size in the z-direction
variable xmax equal 16.06263014623748 # size in the x-direction
variable ymax equal 47.45554551866185 # size in the y-direction
variable zmax equal 41.07662661467842 # size in the z-direction
variable time_step equal 0.001 # time step in pico seconds
variable time_eq equal 1000 # time steps for the equlibration part
variable time_shock equal 2000 # time steps for the piston
variable vpiston equal 0.01 # piston speed in (km/s) multiply by ten to obtain (A/ps)
variable Nevery equal 10 # use input values every this many timesteps
variable Nrepeat equal 5 # number of times to use input values for calculating
variable Nfreq equal 100 # calculate averages every this many timesteps
variable deltaz equal 3 # thickness of spatial bins in dim (distance units)
variable atomrate equal 100 # the rate in timestep that atoms are dump as CFG
variable tdamp equal “v_time_step100" # DO NOT CHANGE
variable pdamp equal "v_time_step1000” # DO NOT CHANGE
variable Up equal “10*v_vpiston”
#variable Up equal -0.005
timestep ${time_step}
# ------------------ Create Atoms ---------------------
read_data shifted.lmp
neigh_modify one 10000
region bulk block {xmin} {xmax} {ymin} {ymax} {zmin} 39
region piston block {xmin} {xmax} {ymin} {ymax} 39.5 {zmax}
group bulk region bulk
group piston region piston
# ------------- Define Interatomic Potential -------------
pair_style vashishta
pair_coeff * * SiO.1990.vashishta Si O
thermo 10
thermo_style custom step pxx pyy pzz lx ly lz temp etotal
dump d all custom 100 dd.dump id type x y z
compute myCN bulk cna/atom 1
compute myKE bulk ke/atom
compute myPE bulk pe/atom
compute myCOM bulk com
compute peratom bulk stress/atom NULL
compute vz bulk property/atom vz
velocity piston create {stemperature} {myseed} rot yes dist gaussian
fix equilibration bulk npt temp {stemperature} {stemperature} {tdamp} iso 0 0 {pdamp} drag 1
variable eq1 equal “step”
variable eq2 equal “pxx/10000”
variable eq3 equal “pyy/10000”
variable eq4 equal “pzz/10000”
variable eq5 equal “lx”
variable eq6 equal “ly”
variable eq7 equal “lz”
variable eq8 equal “temp”
variable eq9 equal “etotal”
fix output1 bulk print 1 "{eq1} {eq2} {eq3} {eq4} {eq5} {eq6} {eq7} {eq8} {eq9}" file run.out screen no
thermo 10
thermo_style custom step pxx pyy pzz lx ly lz temp etotal
run {time_eq}
unfix equilibration
unfix output1
# --------------------------- Shock ----------------------------
change_box all boundary p p s
reset_timestep 0
fix 1 all nve
velocity piston set 0 0 v_Up sum no units box
fix 2 piston setforce 0 0 0
#fix move_pist piston move linear 0.02 0 0 units box
#fix ff piston setforce 0 0 0.000
#velocity piston2 set 0 0 0.000 sum no units box
compute c1 bulk chunk/atom bin/1d z lower {deltaz} units box fix density_profile bulk ave/chunk {Nevery} {Nrepeat} {Nfreq} c1 density/mass file denz.profile
variable temp atom c_myKE/(1.5*8.61e-5)
fix temp_profile bulk ave/chunk {Nevery} {Nrepeat} ${Nfreq} c1 v_temp file temp.profile
variable meanpress atom -(c_peratom[1]+c_peratom[2]+c_peratom[3])/3
fix pressure_profile bulk ave/chunk {Nevery} {Nrepeat} ${Nfreq} c1 v_meanpress file pressure.profile
fix velZ_profile bulk ave/chunk {Nevery} {Nrepeat} ${Nfreq} c1 c_vz file velocityZcomp.profile
variable eq1 equal “step”
variable eq2 equal “pxx/10000”
variable eq3 equal “pyy/10000”
variable eq4 equal “pzz/10000”
variable eq5 equal “lx”
variable eq6 equal “ly”
variable eq7 equal “lz”
variable eq8 equal “temp”
variable eq9 equal “etotal”
variable eq10 equal “c_myCOM[3]”
fix shock bulk print 10 “{eq1} {eq2} {eq3} {eq4} {eq5} {eq6} {eq7} {eq8} {eq9} {eq10}” file run.${stemperature}K.out screen no
thermo 1
thermo_style custom step pxx pyy pzz lx ly lz temp etotal c_myCOM[3]
thermo_modify lost ignore flush yes
run ${time_shock}
undump d