>I am new to LAMMPS. I am looking for help with regards to forming a
nanocluster of SiO2 inside a box of carbonate molecules. For now I used
Material Studio to form the cluster, used the msi2lmp tool to get the
moltemplate data file. For less number of molecules its easier to add this
data in my Carbonate data file, change the co-ordinates etc according to
requirement. But I don't see a easy way to do it for larger number of
molecules(around 5000).>I couldnt not find any option to create a nanocluster data file for
moltemplate. This question is more of Moltemplate, but I ll be very
thankful if anyone knows any best and easier way to get the nanocluster
data along with my carbonate data file (specially the co-ordinates).
Thank-you!--
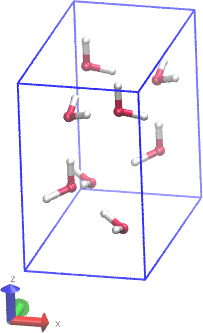
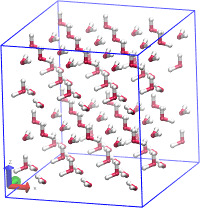
I am attaching two images. One show's the SiO2 cluster I am trying to make.
and the other is the entire simulation box along with the cluster at the
center. Its a amorphous alpha crystal of SiO2.
I ll give you a brief idea about what I am trying to do.
First there is a pure eutectic of Li2CO3 and K2CO3. Then we add 1% SiO2 nano
particles in this pure mixture and see its effect on the structural changes.
I am not able to create the nanocluster in moltemplate. I do not have
enough knowledge of how to make it.
Hope this helps,
Dear Vidula
First of all, the DATA file you sent me is a raw LAMMPS data file
(read by "read_data"), not a moltemplate (.LT) file. (You can convert
data files into moltemplate files using the "ltemplify.py", script,
but I'm not sure if it would be necessary to use moltemplate in this
case.)
For a description of DATA files, go here:
For an example of a moltemplate file, go here:
http://www.moltemplate.org/examples/nanotube+water/spce.lt
Secondly, moltemplate requires a bash shell environment, and I noticed
you are using windows. You will need to install mingw or cygwin if
you plan to use moltemplate in windows (see rant below).
Thirdly, your SiO2 crystall is full of bonds. Do you need these bonds?
I was going to suggest how to build your system using moltemplate and
packmol. Then I looked at your drawing more carefully and I noticed
that your SiO2 crystal contains bonds between atoms in different unit
cells. Moltemplate can make crystals containing bonds, but it is not
smart enough to connect nearby atoms in neighboring unit cells
automatically. Assuming that you actually need these bonds to keep
the crystal stable (or prevent it from dissolving), then I would not
use moltemplate.
However, if you are using a pair_style like REAXFF with parameters for
SiO2, then you don't want these bonds. In that case, read on...
Also, I wanted to mention the issue with the material studio. The data file
that I get from msi2lmp tool has repeated molecules of Si and O. I mean in
atom type there are 2 types of Si and O. With this all the other types, viz
bond-coeffs, angle coeffs etc increases where as actually I should have only
single types of Si and O, two band coeffs and two angle co-effs. I cannot
just manually remove the extra ones, because its hard to figure out which
refers to which molecule.
Fourthly, you said that all of the atoms in your data file already
have the correct coordinates and charge, but the wrong type.
(Question: Are you SURE these are the wrong type? Just because there
are only two elements in your simulation, Si and O, does not mean
there should only be two types of atoms. Perhaps the other atom types
correspond to Si and O atoms with different bonding states and
different charge?)
Assuming you are correct, and there is a problem with the atom types
in your DATA file, in that case the easiest thing to do would be to
use a text editor to cut out the "Atoms" section, and then use awk (or
excell) to change the 3rd column of this file to the desired atom
type. For example if you want to change the third column of a text
file from 3 or 4, to 1 and 2 respectively, type this into the bash
terminal:
awk '{if ($3==3) {$3=1} else if ($3==4) {$3=2} print $0}' < old_data_atoms.txt
Learning "awk" is cool. But, for a system this small, you could
accomplish the entire task with a text editor, changing the atom types
manually. If you are sure the atom types are incorrect, try doing
that.
----- building the system using moltemplate -----
This will require an investment of time and effort.
Side note: for this to work, you will need a "bash shell", so if you
are using windows, you will need to install mingw or cygwin or
linux/mac. (I think this is one of many examples which illustrates
why if you are serious about doing any kind of computational work, I
would move away from a graphical windows/DOS environment. Even if you
don't understand how they work, you will want access to classic tools
like a compiler, awk, python, sed, and get familiar with a text editor
which is not word. Learning not to fear these fundamental tools is an
investment in your future. Forgive me if you already are familiar
with them.)
If you want to start from scratch using moltemplate, I would use this strategy.
a) I recommend that you create a "unit cell" object containing all of
the atoms (and bonds, angles, etc.) which would be contained in a
minimal unit cell of SiO2. Again bonds between atoms in different
unit cells are not allowed, so you would need a force-field which does
not need these bonds, like reaxFF.)
b) Use the new command to create a 3-dimensional array of unit cells
(see "ice_rect8_crystal_3x2x2_LR.jpg"). This crystal will have a
rectangular or orthorombic shape by default, but you can use the
"delete" command to remove unit cells near the boundary to give it a
rounder shape. (You may need to change the partial charges on the
boundary atoms, and make other customizations. Moltempate does not do
this for you, but it sounds like msi2lmp does.)
c) Surround the crystal you created using a 3-dimensional array of
carbonate molecules (also created using the "new" command), and then
use one or more "delete" commands to carve out a hole in the middle
where the the SiO2 crystal will be located. For more details how to
do both of these tasks, see this post on the LAMMPS mailing list:
d) Then run a simulation to move the carbonate molecules around
randomly. Alternately, you can also use PACKMOL to replace steps 3
and 4.
http://www.ime.unicamp.br/~martinez/packmol/
Then you would use "moltemplate -xyz" to read the coordinates which
PACKMOL generated, but you first have to extract the coordinates of
the SiO2 crystal you created in step 2 and feed these coordinates to
PACKMOL so that it knows not to put the carbonate molecules at this
location.
Cheers
Andrew