Hello, everyone. I am utilizing the ICET to deal with a binary(Al-Cu) system MC simulation. I have trained a ce model by iteration which is the same to the Unicle. But I found some problems really making me confused. I am looking forward to someone who may help to give perspectives to these questions. My questions are as follow:
1.when training a ce model, the nmsd(Normalized mean-squared displacement) is a significant value to determine whether a structure should be added into the structure container or not (DOI:10.1103/PhysRevB.96.014107), higher nmsd means greater distortion after structure optimization, which may contribute to failure of the ce model. So at the beginning, I only choose stuctures with small nmsd adding into the structure container. But in the iteration process(using the ce model to predict the whole configuration space, if there are structures with lower enrgy than the structures in training set, add them to the structures container, then train the ce model agian;iterate this process untill no structures with lower energy was found), I found that some structures with lower energy but larger nmsd, if I add these structures into sc, it may do bad to the ce model(in fact, predictions on these structure with larger nmsd will have a bigger errors relative to the reference energy calculated by dft(the error for structures with higher nmsd can reach 60mev), and this is reasonable because clusters of strutures with bigger nmsd may change after structure optimization if a bigger distortion happened); if I don’t add these structures into sc, the iteration process will never come to an end, and this means some potential base structures didn’t include into the training set, which may lead to some potential mistakes.
So is there a method helping to determine whether these structures shoulde be added into sc or not?
2.How to judge whether a ce model is reasonable or not?
I utilize the ce model I trained to predict the whole configuration space, it seems all right to almost all the structures except two pure components structures(Al and Cu) and some structures with larger nmsd. For instance, the reference energy(Formation energy) for single Al and single Cu are 0,but the preditced energy for these two are 13.95mev and 14.98mev respectively, which is much more bigger than the training error(3.9mev) and validation scroce(testing error 4.68mev). So confusing! As I descripted in the first question, structures with small nmsd usually correspond to a accurate prediction, however, the nmsd values of these two pure component structures are 0 while the prediction are not accurate as usual, which confuses me a lot.
I wonder that whether this condition is normal or not? if it is not, could you please give me some advices to figure out the uncovered mistakes?(Though I think bigger error on the two pure componet structure may not affect the MC simulation since the reference energy for these two structures are almost the biggest among the configuration space, it still make me uncomfortable, hoping someone can give a more convincing idea)
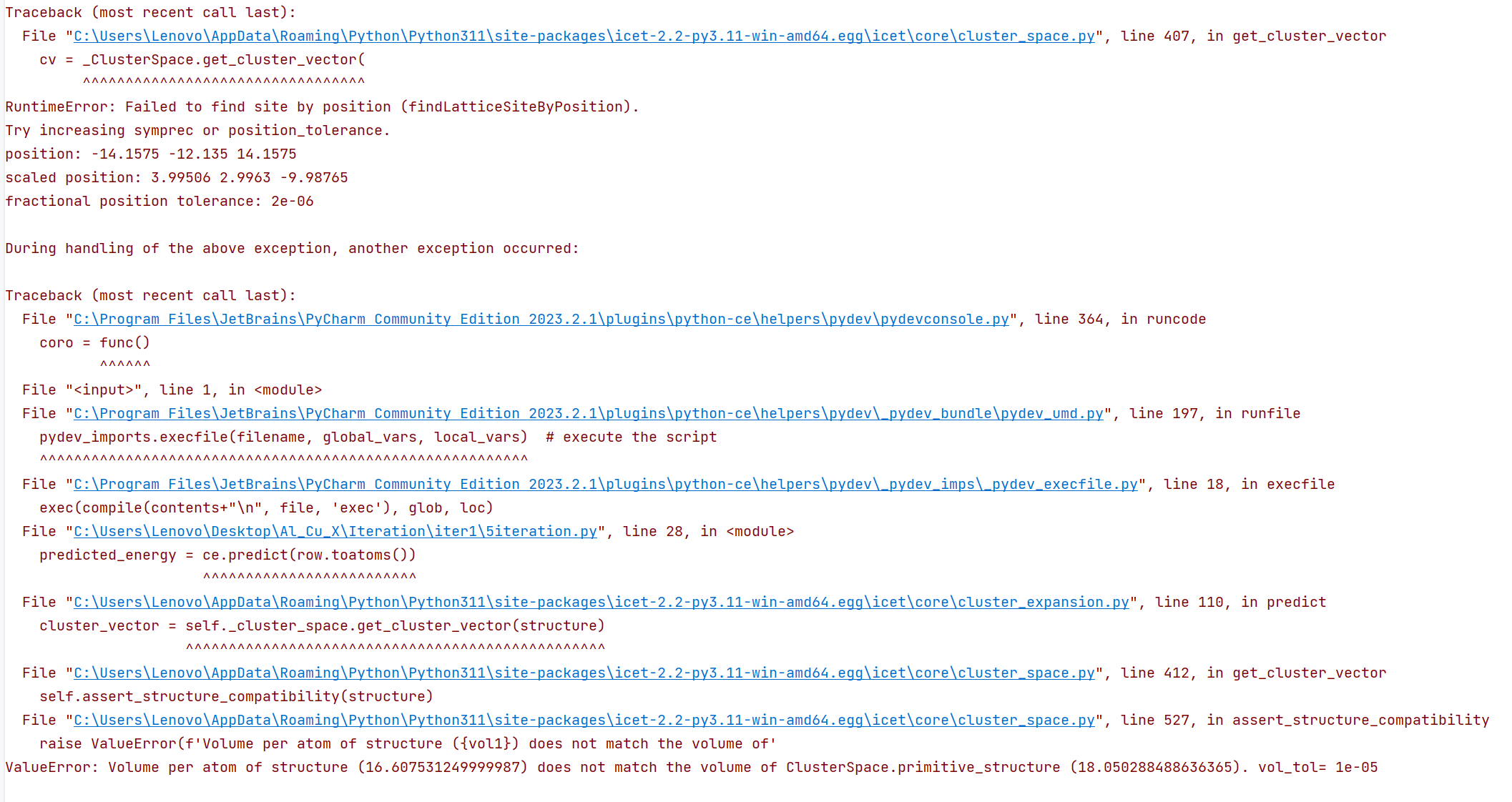
3.In ICET tutorial, there is a portion instructing how to analyse ECIs, Analyzing ECIs — icet documentation (materialsmodeling.org).
In source code, there is a method named ‘orbits_as_dataframe’ , however, I went through the ClusterExpansion module and found no method named ‘orbits_as_dataframe’.
Could you please tell me where I can find this method?
4.When the kinds of elements increase, the number of enumarate structures will explode, which makes it unavailable to claculate all the structures, instead we choose some of the enumerate structures as initial training set and gradually perfect the training set by iteration. Do you think it is necessary to utilize some principles to determine the initial training set? For example, choose strutures which are the most different by Fingerprint of structures?
From the point of my view, Cluster Expansion could be regarded as a machine learning process(regression), then the training data containing more information of configuration space will be better. An initial training set with the most different structures should contain more information of the configuration space. Picking out the most different structures colud be implemeted by fingeprint.
I wonder that have you thaught of this question, is it a necessary process to conduct? Or from the point of your view, what should be the most ideal method to train a ce model with high reliability?
Many thanks and best wishes, looking forward to your reply!
Rocky