Dear all
I am using reax/c during the NEMD simulation with wall/reflection.The system is RDX crystal.I have some questions about the reax/c:
- how to calculate the bond energy, such as C-C, C-H, C-N et al. bong energy?
I saw the manual and found the compute bond/local command may calculate the bond energy.
compute 2 all bond/local dist engpot
dump 1 all local 1000 tmp.dump index c_2[*]
ERROR: Compute bond/local used when bonds are not allowed.
How to modify it?
- how to subtract the center-of-mass velocity?
compute myTemp all temp/com
compute RDXX hot chunk/atom bin/2d x 0 5 y 0 5 units box
fix 6 hot ave/chunk 100 1 1000 RDXX vx vy vz temp density/mass bias myTemp norm sample file tmp_RDX.out
The temp in fix command is the real temperature, is it right?
- how to calculate the real press?
In lammps, the press is computed by the formula
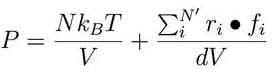
compute peratom hot stress/atom NULL
variable meanpress1 atom -(c_peratom[1]+c_peratom[2]+c_peratom[3])/3
fix 7 hot ave/chunk 100 1 1000 RDXX v_meanpress1 norm sample file tmp_RDX_press.out
Is the v_meanpress1 in fix command the real press?