Dear LAMMPS USERS,
I attached two pictures that first one is my model(Carbon walls and SPC/E water) and the second one is SPC/E and nanotube model presented in the moltemplate home page.
Could anybody tell me whether my SPC/E water configuration is wrong?
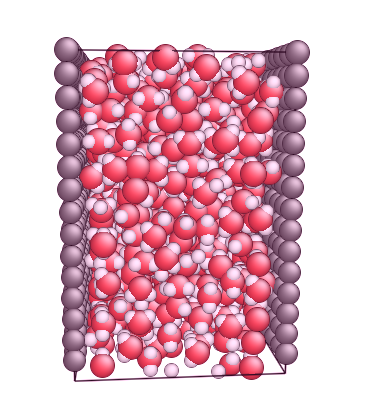
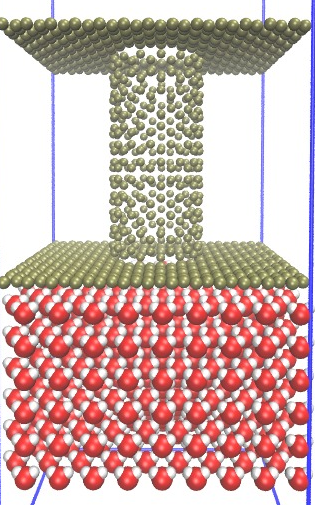
How are we supposed to be able to tell from a picture if your configuration is right or wrong? The right-hand figure was probably done with water starting in a crystalline arrangement, so you shouldn’t read anything into that if you built your molecules using a liquid-state configuration.
But otherwise, we can’t say anything. You have to check if you used the correct geometry, charges, etc.
—AEI
Dear Ahmed,
I made the my model (first attached file) through randomly inserting water molecules into the box.
My main question is that if the SPC/E water model arrangement accepted by lammps MUST be as similar as the 2nd pic or the attached file?
because I have read from the moltemplate (http://www.moltemplate.org/) that it mentioned that " try to insure the waters are spread uniformly throughout the box ". But my model is not uniformly distributed.
Actually, I got some nan errors when I calculated the pressure and potential via thermo_style command. Some lammps experts conveyed that the overlapping or bad initial configuration might be caused such errors.
So, I want to make sure about my model.
Thanks in advance
Dear Ahmed,
I made the my model (first attached file) through randomly inserting water
molecules into the box.
My main question is that if the SPC/E water model arrangement accepted by
lammps MUST be as similar as the 2nd pic or the attached file?
because I have read from the moltemplate (http://www.moltemplate.org/) that
it mentioned that " try to insure the waters are spread uniformly throughout
the box ". But my model is not uniformly distributed.
Actually, I got some nan errors when I calculated the pressure and potential
via thermo_style command. Some lammps experts conveyed that the overlapping
or bad initial configuration might be caused such errors.
So, I want to make sure about my model.
you are missing the point. nobody can tell this from a picture (unless
you made an error of epic proportions).
you have to look at the actual data and/or visualize it. also you have
to factor in your choice of boundaries; with periodic boundaries
overlaps may be not so obvious if the overlapping atoms are on
different sides of the box and only overlap with the periodic image of
each other.
axel.
Dear Majid
It sounds like you need to minimize your system. The picture shown
on the moltemplate web page:
http://www.moltemplate.org/images/nanotube+water/water_side_nopbc.jpg
..is just one of many ways of arranging water molecules. In this
particular example, the water molecules were arranged in a hexagonal
lattice, but that detail is not important (although it might make the
example more confusing).
What is important is that you should minimize your system before
you attempt to run the simulation. This is true regardless of how you
arrange the molecules. The LAMMPS input script for the nanotube+water
example invokes the "minimize" command before it invokes the "run"
command:
http://www.moltemplate.org/examples/nanotube+water/run.in.nvt
(An unimportant detail: It also turns "fix shake" off and on, because
"fix shake" is currently incompatible with "minimize".)
Sometimes the default "minimize" command fails, and you have to
use a slower method. It's been a while since this happened, but I
think I remember that I was able to get good results using something
like: "min_style quickmin" and "min_modify dmax 0.02".
http://lammps.sandia.gov/doc/minimize.html
http://lammps.sandia.gov/doc/min_style.html
http://lammps.sandia.gov/doc/min_modify.html
You can also use the dump command before the minimize command, for example:
http://www.moltemplate.org/examples/martini_DPPC_bilayer/run.in.min
This will create a trajectory file you can visualize in VMD. (See
sections 4.3 and 4.5 of the moltemplate manual.)
Good luck
Andrew
Dear Andrew, Axel and Ahmed,
Thank you very much for your valuable time and comprehensive answers to my trivial question.
Thanks in advance