Dear Dr. Axel Kohlmeyer and LAMMPS Users,
Hello All. I am simulating a P3HT polymer using coarse-grained (CG) molecular dynamics.
The system of P3HT consists of three types of CG particles, Type 1, 2 and 3. Type 1 atoms make up the backbone monomers of P3HT whereas Type 2 and 3 coarse-grained the hexil side chains.
I would like to tabulate the angle potential using the command angle_style table for angle between CG particle types 1,1 and 2 and between types 2, 1 and 1. (The 3 particles types are ordered linearly within the angle such that the particle type in the middle corresponds to the center particle in accordance to how the three particles are ordered in the data file. In the data file, the particle types are listed as corresponding atom ID’s.)
However, I would like to tabulate angle potential for particle 1,1,2 and for 2,1,1 which consists of two local minimums thereby possessing two metastable equilibrium angles as shown in the below figure.
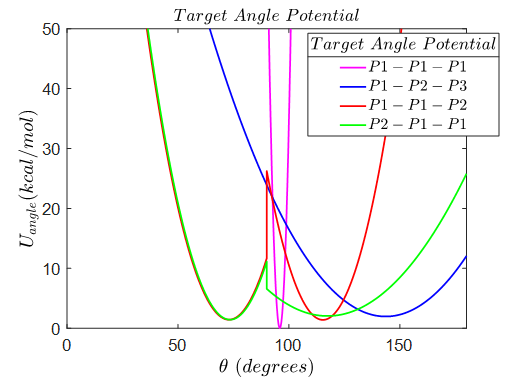
Observing the table that I need to create when I tabulate potential, I see that I must specify the tabulated angular displacement, energy and force. I also noticed that the second line of the table must be in the format: N 181 FP 0 0 EQ 90.0 where I only have one space for specifying the equilibrium angle. I understand that FP and EQ are optional but if I do not specify EQ, the equilibrium angle is set to 180 degrees. Is there a way in LAMMPS to have the table to enlist two potential equilibrium angles (e.g. 70 and 120 for angle 1,1,2 and same for angle 2,1,1)? Thank you so much for your time! Sincerely, Masato Koizumi