Hi there,
I am trying to figure out if I am using the optimal settings with LAMMPS and OpenMPI while running on a supercomputer cluster to optimize the simulation speed of my system.
I realize I am using a computationally expensive interaction potential (reaxFF), but in a small system test (128 Carbon atoms, density of 2.0 g/cc, 157000 timesteps, 10 simulation runs) I still believe there is much improvement to be made with respect to how long it is taking to finish the runs.
For example, speciyfing 1 node and ranging the number of CPUs I find the following relationship between number of CPUs and total runtime:
1 CPU = 6.25 hours
2 CPU = 5.75 hours
4 CPU = 5.5 hours
8 CPU = 3 hours
12 CPU = 2.25 hours
16 CPU = 3.05 hours
24 CPU = 5.5 hours
32 CPU = 3.25 hours
Below I paste the modules I am loading and the general pseudocode of my script (.sh) which I am submitting to a ‘slurm’ based scheduler on the cluster:
START for loop (1 to 10): generate random seed number from 1 to 10 million change the dump file name based on the seed number generated rewrite the input textfile based on the randomly generated seed value (only changing create_atoms) run for 157000 timesteps srun lmp < in.txt
END loop
Note that I do not use the ‘clear’ command anywhere in my LAMMPS in.txt file and I understand that using the same directory for different seed runs results in the same initial configuration.
I am wondering if this for loop (which was the most intuitive way for me to repeat random seeds runs) is tanking my code performance or if I am missing a module specifier for OpenMPI. I can happily share the output textfile that I recieve from the cluster.
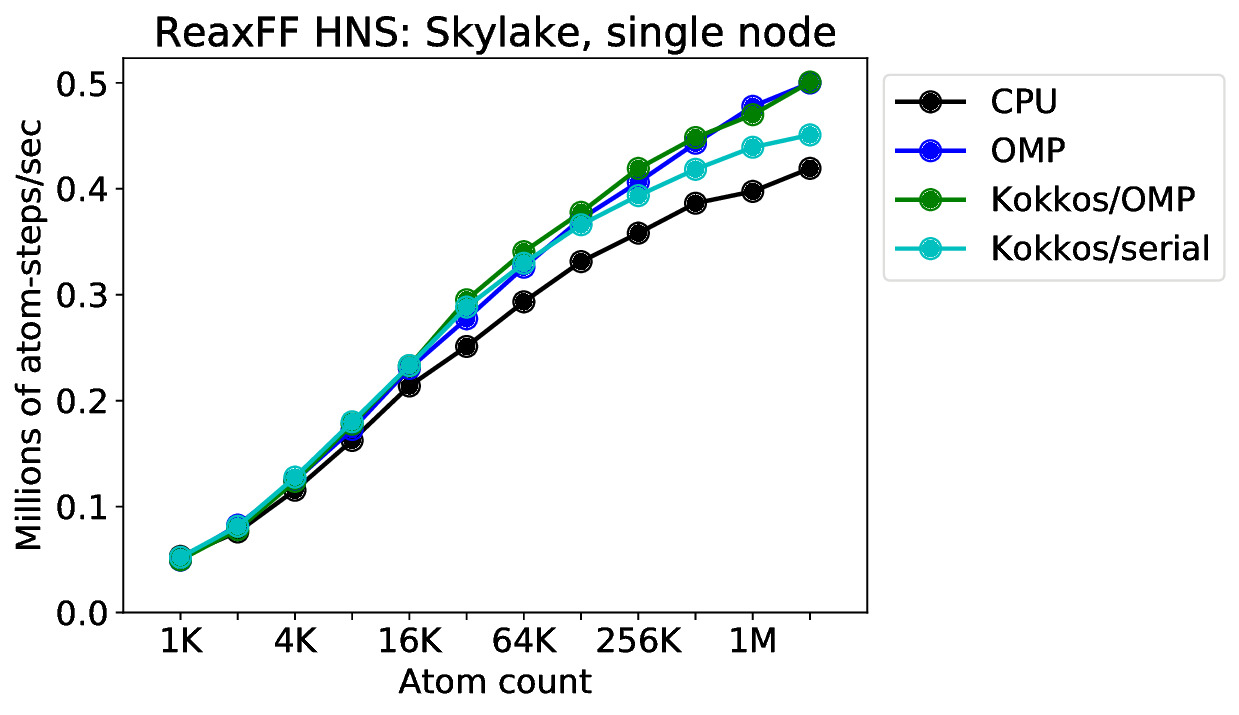
With 128 atoms and 32 MPI ranks, you only have 4 atoms per MPI rank. ReaxFF is moderately expensive but still cannot strong scale to that extreme. The ratio of ghost halo exchange vs simulation volume is going to be huge. In other words, the simulation is nearly all MPI communication and almost no computational work. This is explained well in this paper: https://doi.org/10.1002/jcc.20267. LAMMPS uses a “half-shell” method, and would need a “neutral-territory” method instead to further scale to the extreme. As shown in the plot below for an Intel Skylake CPU, you need ~2 million atoms to saturate the CPU with 36 MPI ranks:
I see, thank you for the explanation. If I understand correctly, even if I were to expand my system to my target of 2000 atoms, there is almost no benefit to using an increasing number of CPUs.
Is there any explanation for why 12 CPUs seems to show the fastest completion time (would this imply a lower communication time with 12 than for any of the other number of CPUS)?
I think there would certainly be some benefit to using 2000 atoms in terms of total atom-steps/s performance–you are making the system nearly 16x larger than 128 atoms, so you can scale out to more MPI ranks. As you strong scale the system, the time spent in computing forces goes down with more MPI ranks since each rank has fewer atoms, but the time spent in communication goes up. So there is a crossover point = 12 MPI ranks in your case where the cost of increased communication outweighs the benefit of reduced computation.
As a final question, could my use of a for loop for triggering consecutive simulations with differing seeds capable of slowing down the simulations (or would it be an insignificant slow down)?
The loop should have negligible overhead since you are running for hours. We normally use “Loop time” from the log file to measure performance rather than overall time–overall time includes MPI and LAMMPS initialization not in “Loop time”, but that should also be small compared to hours of run time.
If you are looking for extra parallelism, you could use a uloop style variable for the loop and do a multi-replica run. That way you would do multiple runs with different loop iterations concurrently on the different partitions.
That sounds very promising especially for the small systems I intend to simulate. Is there another page on the documentation that might provide an example of where to include the uloop command in my in.txt file? Or is this page: variable command — LAMMPS documentation the only reference?
Also if I could ask a very basic question, is a partition the same as a CPU in my case using OpenMPI?
No, it can be a group of MPI processes. With -partition 8x4 you would run on 8 partitions of 4 MPI processes each, and thus would have to launch LAMMPS with mpirun -np 32.
You didn’t mention this so apologies if you are already aware – but do note that on a supercomputer cluster you will almost certainly have a fixed number of “service units”, or SUs, with which to run your simulation. One service unit is one CPU running for one hour (possibly multiplied by some proportionality constant).
So you should not only measure the walltime of your calculation, but also the number of CPU-hours used. If you look at the results table in your first post you will see that your 1 CPU run, even though it takes the longest, only uses 6.25 CPU-hours; your 12 CPU run, while being the fastest, uses 30 CPU-hours. So budget your CPU-hours and don’t just aim for the fastest possible simulation (and burn through your resources). Also, you should run these scaling tests on your actual system; trying to predict a large system’s behaviour from a small system’s behaviour is not very productive.
Finally, talk to your cluster administrator. Make sure that your executable is properly compiled against their MPI build, and make sure to ask about the appropriate settings for grouping MPI threads on nearby cores (the --distribution directive in SLURM).
Just for posterity and for other users who might find this post.
I managed to successfully modify my scripts following the documentation listed above in addition to using this forum post: Question about how to multiple doc page