I’m running a very simple system of 4000 spheres with embedded dipoles, intended to be a coarse grained water simulation. The log file is attached. In order to thermostat the rotational degrees of freedom, I thermostat the system using “fix langevin omega yes” for 20000 timesteps (10fs per timestep) and then run an NVE simulation for 100000 timesteps by simply unfix-ing the thermostat. During the thermostatted portion of the simulation, the temperature appears well thermostatted and the total energy seems to be sensible. However, over the first 100-200 timesteps of the NVE potion of the simulation, ie without any thermostatting applied, the total energy of the system increases and the system temperature falls to new stable values where they remain for the rest of the NVE portion.
I have defined a compute (compute all temp/sphere dof all) which includes the spheres’ 6 degrees of freedom for the temperature calculation and used that compute for the temperature in both thermo_modify and fix_modify. This has reduced the increase in total energy at the beginning of the NVE run that I was previously seeing but there still is an increase. If I run the system without dipoles (ie I don’t include the set all dipole/random line and just leave the default 0 implied by create_atoms) I don’t get the increase in total energy at the beginning of the NVE portion.
I’ve tried various different timesteps (1.5fs - 15fs) and I see the same energy increase over a similar time for each. Can anybody shed any light on what I’m getting wrong - I suspect there’s a default I need to change but I’ve checked the manual thoroughly for all the commands I’m using and I can’t find it.
Yes (at least, I hope so!)
I define a compute, all_temp which is temp/sphere dof all. I then use thermo_modify (after the thermo_style line!) to make sure that the temp returned by thermo_style is calculated using that compute. I then use thermo 100 to return temp and etotal every 100 steps throughout the thermostatted and non-thermostatted portions.
The input script is attached in case I’m not being clear…
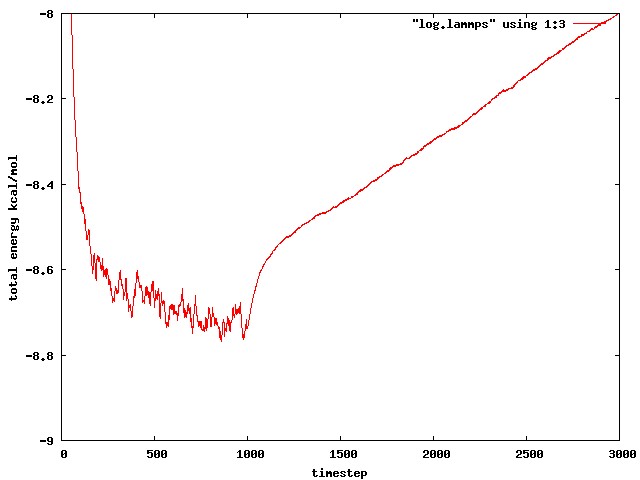
I’m sorry, I should have said in my initial email, I get a similar energy jump using dipole/cut. Its slightly harder to see because using the cut potential does not conserve the energy so well in the NVE portion of the simulation. I have run a short simulation (1000 steps NVT, followed by 1000 steps NVE) using dipole/cut. Hopefully you can see from the attached jpeg that the total energy increases very fast at the beginning of the NVE portion, much faster than the energy drift later in the NVE portion. I have also attached the input script for this dipole/cut simulation.
the issue here is not about the energy conservation generally - I agree that using a straight cutoff the total energy in an NVE run should steadily increase (as it does in this case).
The problem is that the energy shows an unexpected jump at the very beginning of the NVE run (step 1000 in the diagram attached by Sophia in her previous message), before settling on a steady increase.
Using the shifted-force version (where a better energy conservation is expected), the same issue shows up as an anomalous jump at the very beginning of the NVE run, which you can see in the first three lines of this portion of the log file that Sophia attached to her first message:
Why does the total energy initially increase before settling on a constant value (-7.27[…])? It should be fluctuating about a constant value from the very beginning.
In fact, if the dipoles are removed, this issue also disappears, i.e., the energy steadily increases using the cut potential (no jump), or immediately settles on a constant value using the sf potential.