I’m working on some sputtering simulations. This script is giving reasonable results. Additionally, I’m trying to investigate the chemical reactions/molecules formed when a target is bombarded with Iodine ions (I2). I’m using this command: fix 5 all reaxff/species 1 2 10 species.out cutoff 1 1 0.40.
# Sputtering Iodine on C Case sputter
# 3D simulation parameters
dimension 3
units metal
atom_style charge
boundary p p s
# Simulation box dimensions
variable xlo equal 0.0
variable xhi equal 90.0
variable ylo equal 0.0
variable yhi equal 80.0
variable zlo equal 0.0
variable zhi equal 90.0
# Delta between insert area and box
variable len0 equal 0.1
# Full length minus delta
variable len1 equal ${xhi}-${len0}
# Half length
variable xhi2 equal ${xhi}/2
variable yhi2 equal ${yhi}/2
variable zhi2 equal ${zhi}/2
processors * * 1
#region simbox block ${xlo} ${xhi} ${ylo} ${yhi} ${zlo2} ${zhi2} units box
#lattice diamond 3.5
lattice custom 3.5 a1 2.13 -1.23 0.0 a2 2.0 -1.23 0.0 a3 0.0 0.0 6.71 &
basis 0.0 0.0 0.0 basis 0.5 0.5 0.5
region simbox block ${xlo} ${xhi} ${ylo} ${yhi} -1.0 40.0 units box
create_box 2 simbox
region layer block ${xlo} ${xhi} ${ylo} ${yhi} ${zlo} ${zhi} units box
#create_atoms 1 region layer
create_atoms 1 random 1000 12345 layer
# Define masses
mass 1 12.0107 #Carbon (C)
mass 2 253.8 #Iodine (I)
region fixed block INF INF INF INF 0.0 0.1
group fixed region fixed
group deposited subtract all fixed
# Define Interatomic Potential
pair_style reaxff NULL
#pair_style reaxff NULL lgvdw yes
#pair_style reaxff amp_control
pair_coeff * * ffield_1 I C
# Define charge equilibration fix
#fix 1 all req/reax 1 0.0 10.0 1.0e-8 reaxff
fix 1 all qeq/reaxff 1 0.0 10.0 1e-6 param.qeq
neighbor 0.3 bin
neigh_modify every 2 delay 6 check yes
# Set the force on atoms in fbot to Zero
#fix bl fixed setforce 0.0 0.0 0.0
minimize 1.0e-5 1.0e-8 100 1000
reset_timestep 0
compute mtemp deposited temp
#fix 2 deposited nvt temp 200.0 200.0 $(1.0*dt)
fix 2 deposited nve
fix 3 deposited langevin 0.2 0.2 5.0 982434
fix_modify 3 temp mtemp
# Relax the I-C Substrate and thermalize
thermo 100
thermo_style custom step atoms etotal
thermo_modify temp mtemp
dump id all xyz 200 sputter.xyz
# Run
run 1000
# Define sputtering region
region sphere sphere ${xhi2} ${yhi2} 42.0 ${zhi2} units box
#region block block 2.0 23.0 2.0 23.0 40.0 50.0 units box
region insert intersect 2 simbox sphere
# Define group of sputtered particles
group insert region insert
neigh_modify every 1 delay 0 check yes
# time integrate sputtered particles
fix 4 insert nve 1.0
unfix 4
variable eV_to_J equal 1.602176634e-19
variable m_I equal 2.1065e-25
#variable E index 250 350 450 500
variable E index 200 400 600 800 1000
variable nE equal 5
region sputterb block INF INF INF INF 38.0 INF units box
molecule I2 I2.txt
variable E_now equal ${E}
label loop
variable e loop ${nE}
variable i_speed equal sqrt(2*v_E*${eV_to_J}/v_m_I)/100
labelmap atom 2 I2
fix sputter insert deposit 100 I2 200 12345 region insert vz -${i_speed} -${i_speed} target ${zhi2} ${zhi2} ${zhi} units box
fix 5 all reaxff/species 1 2 10 species.out cutoff 1 1 0.40
group incident_ion type 2
group sputtered region sputterb
#---Incoming atoms count---
variable nimpacting equal count(incident_ion)
#---Sputtered atoms count---
variable nsputtered equal count(sputtered)
variable m equal 12.0
#variable E_r equal ${nsputtered}*${nimpacting}*${m}/1.2469e+04
#thermo_style custom step v_nimpacting v_nsputtered v_zhi2 v_zhi2 v_zhi ke v_E_r
thermo_style custom step v_nimpacting v_nsputtered
run 5000
print "Energy = $E"
#group incident_ion delete
#group sputtered delete
# Ignore lost atoms since sputtering atoms can fly out of simulation box
thermo_modify lost ignore
next E
jump SELF loop
This is a sample of what I’m getting:
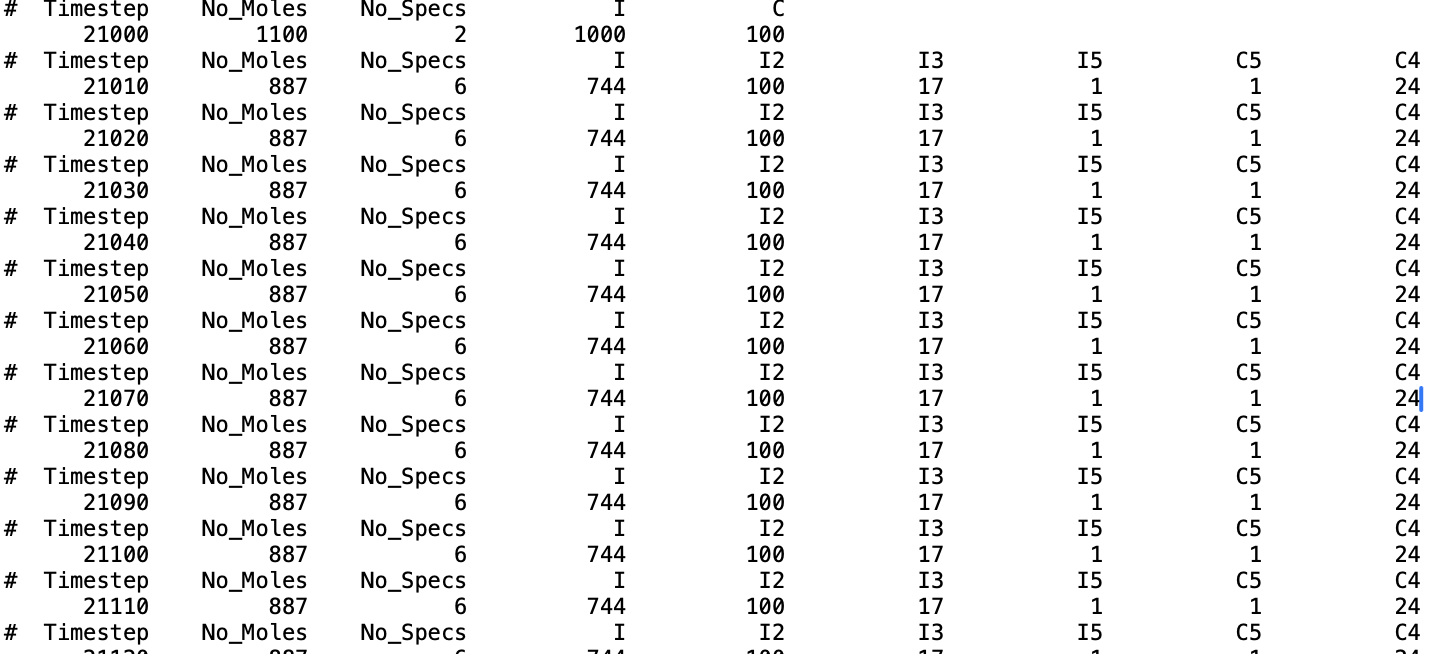
I’m curious why I appears before I2. I’ve adjusted the cutoff value, but I’m still getting the same result. Any help or guidance for better comprehension would be greatly appreciated. Thank you.