I am modelling Alumina using sw three body parameter and receiving error
ERROR: Unknown table style O of angle table file (src/src/MANYBODY/pair_sw_angle_table.cpp:301) Last command: pair_coeff * * sw/angle/table Al2O3.sw Al O
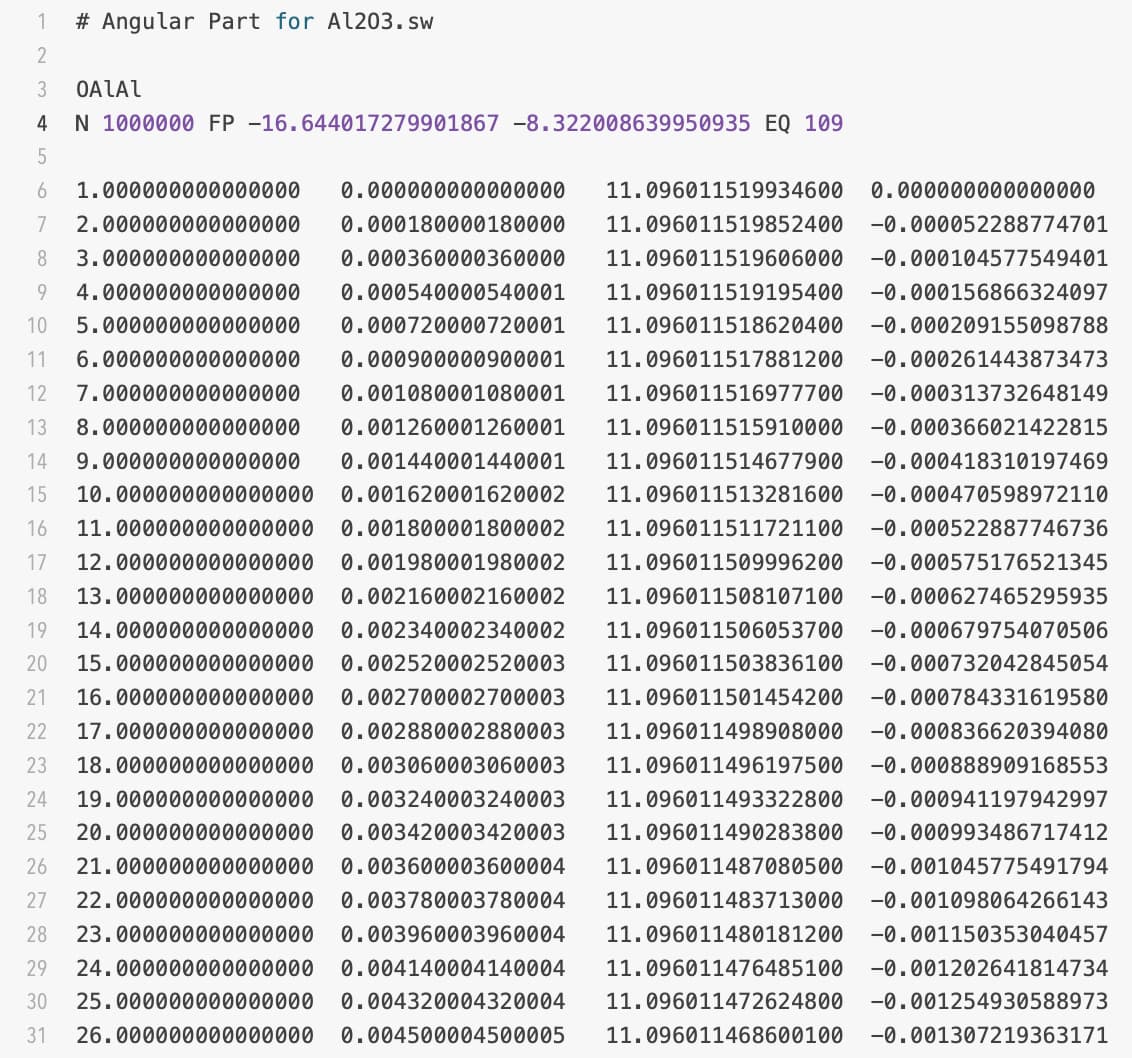
Here are files,
Al2O3 is data file, .sw is potential file, and both text files are tables for angular energy terms.
I hope this is a better method to present my problem.
Your Al2O3.sw file is not correct. You have table files only for the first two entries, but the remaining 6 entries have “0 0 0 0” instead. Hence the error message.
Thank You that fixes the problem,
However I am getting another WARNING: 181 of 181 lines in table incomplete or could not be parsed (src/src/MANYBODY/pair_sw_angle_table.cpp:474)
ERROR: Angle table must range from 0 to 180 degrees (src/src/MANYBODY/pair_sw_angle_table.cpp:359)
Last command: pair_coeff * * sw/angle/table Al2O3.sw Al O
Also I only need AlOO and OAlAl three body parameters, therefore I added a keyword for the rest possible combinations but made values to be zero because of the previous error.
Your geometry is bogus. Just sort the coordinate by x, y, or z and you’ll find that you have multiple atoms with nearly identical coordinates.
In fact, when I add the command “delete_atoms overlap 0.02 all all”, 104 atoms are deleted for being closer than 0.02 angstrom than another atom and only 56 remain. That is a very bad sign.
Yeah Geometry was the problem. Thank you everyone for inputs. It’s running without error now.
Here are files for the paper mentioned in case for future use of anyone else.
Molecular dynamics simulations of α-alumina and γ-alumina surfaces
Hey,
As stated above it worked for another data file. I change the data file according to my needs. and it is still giving me the same error of illegal angle.
I checked my geometry using the overlapping command and 0 atoms got deleted.
Is there anywhere else I should look into.
Additionally, can you help me to know where should I introduce my cutoff of the angular part.
ERROR: Angle_write command before an angle_style is defined (src/src/angle_write.cpp:49)
Last command: angle_write 1 500 table.txt Harmonic_1s
The error in the log file you provide is different from the previous one and therefore requires a different solution that is explicitly stated in the error message.
That is because the pair style does not handle 180 angles well and has no provisions when the argument passed to the acos() function is slightly larger than 1.0 or slightly smaller than -1.0.