Dear all,
I was doing the stretch simulation of borophene at 1K in the NPT ensemble, using fix deform command to stretch. The stretching direction is along the zigzag direction. The potential is sw/mod as stated in the papers and LAMMPS manual.
In the stretching process of borophene, I observed that the angles around the boron atoms are flattened as shown in the figure. This leads to the ultimate strain of borophene much higher than the value in the paper.
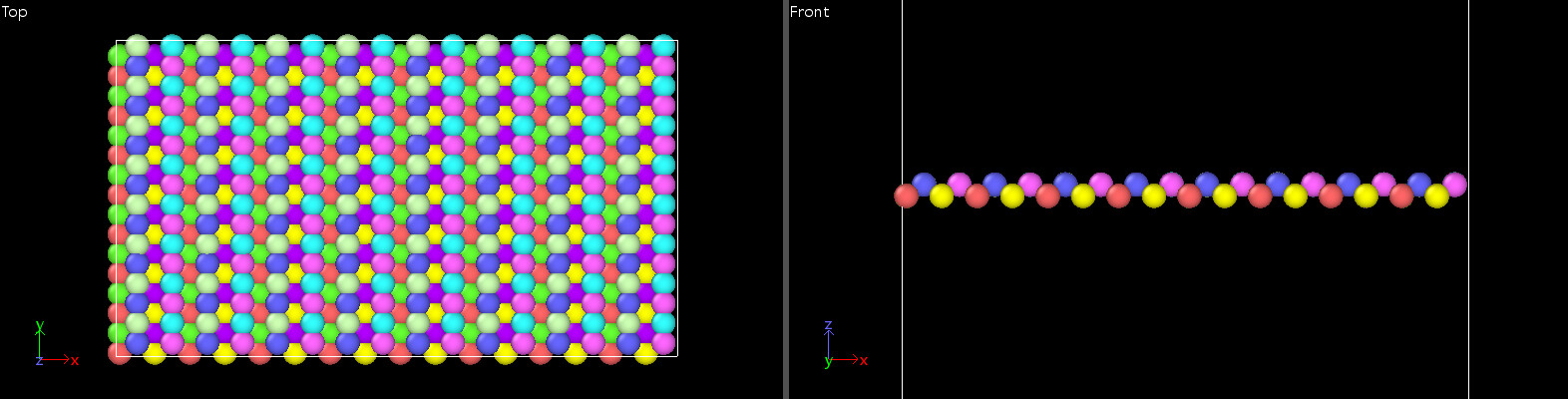
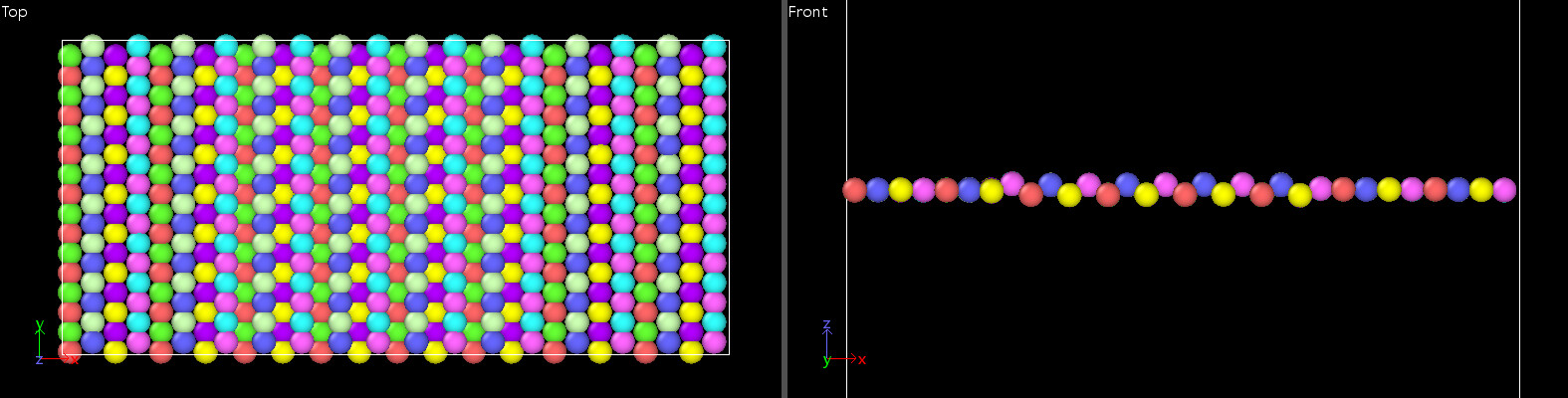
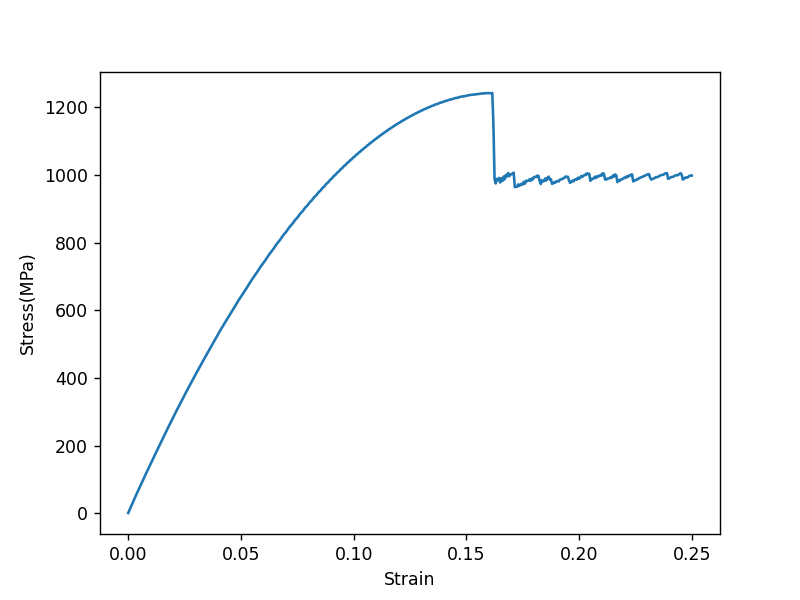
Then I plotted the stress-strain curve of borophene.
The results in this paper are as follows.
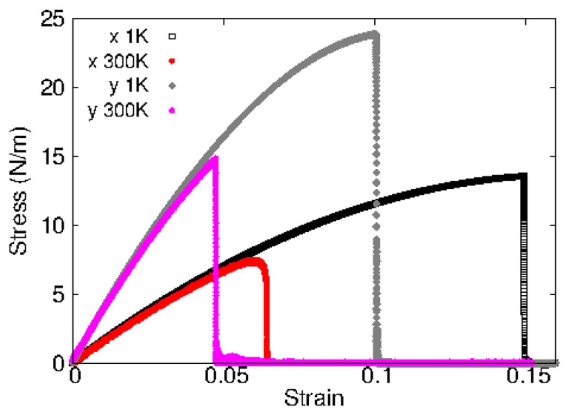
[1] Y.P. Zhou, J.W. Jiang, Scientific Reports, 7, 45516 (2017)
Was there something wrong with my input file?
Here is my input file:
units metal
atom_style atomic
dimension 3
boundary p p p
timestep 0.001
neighbor 0.3 bin
read_data boron.data
replicate 4 4 1
mass * 10.81
pair_style sw/mod maxdelcs 0.25 0.35
pair_coeff * * borophene.sw B1 B2 B3 B4 B5 B6 B7 B8
# minimize
fix rlx all box/relax x 0 y 0
thermo 100
dump dmp1 all atom 10 mini.xyz
min_style cg
minimize 0 1.0e-8 10000 10000
unfix rlx
undump dmp1
reset_timestep 0
variable T equal 1
velocity all create ${T} 123456 rot yes mom yes dist gaussian
fix mynpt1 all npt temp ${T} ${T} 0.1 x 0 0 1 y 0 0 1
thermo 1000
dump dmp2 all atom 1000 relax.xyz
neigh_modify once yes
run 30000
# tension
unfix mynpt1
undump dmp2
reset_timestep 0
fix mynpt2 all npt temp ${T} ${T} 0.1 y 0 0 1
fix dfm all deform 1 x erate 0.001 remap x
variable Lx equal lx
variable Strain equal (lx-${Lx})/${Lx}
variable Stress equal -pxx*0.1
fix out all print 500 "${Strain} ${Stress}" file strain.txt screen no
thermo_style custom step temp press pxx pyy lx ly v_Strain v_Stress
dump dmp3 all atom 500 stretch.xyz
run 250000
Thanks!
Sun