Hi, lammps-users
I’d like to ask the structure of materials after running lammps.
I have tried to calculate the phonon density of state of graphene on Cu with VACF command.
First I make single-layer graphene on Cu and lammps script called it.
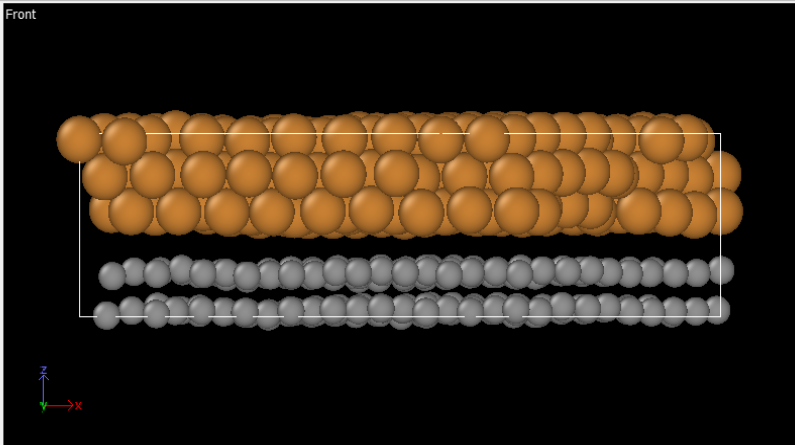
Once I ran lammps and I checked the geometry of materials, graphene layer was not single.
Also, PDOS shape of graphene on Cu was different from literature.
Here is my lammps script and I attached picture and geometry scripts.
I’d really appreciate it if you give some comments.
Thanks,
Jaeyoung Jeong
This is the control script for LAMMPS
echo both
g-cu-4.data (21.6 KB)
Lammps might be confused with the following region commands. Both the Zmin and Zman are same. Try with different values (say 1.48 1.52).
region gra_inter block INF INF INF INF 1.500000 1.500000 units box
Sanjib
Hi, Sanjib
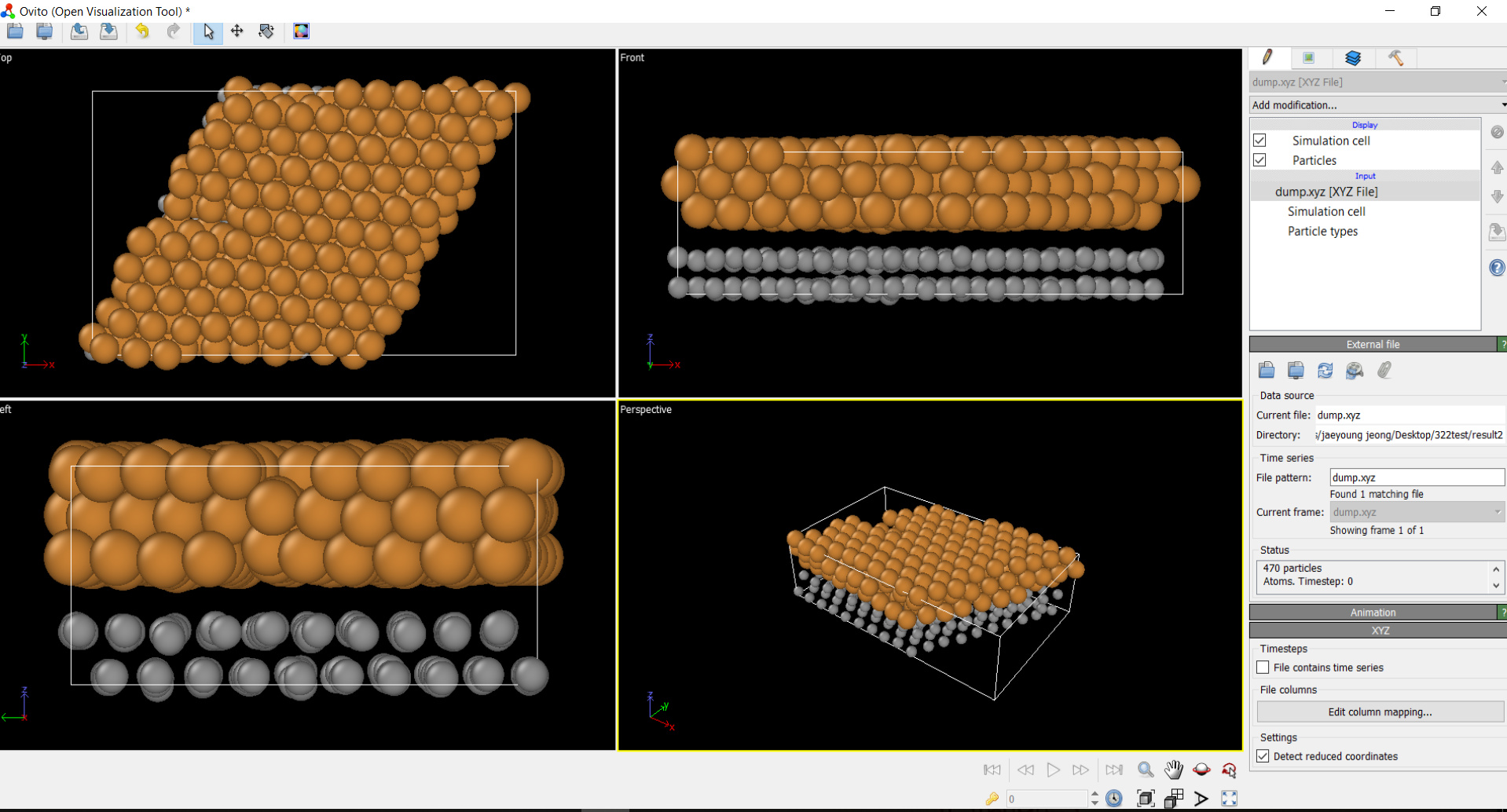
I ran LAMMPS based on your comments as below script but the geometry was the same as attached files.
I wonder where my lammps script was wrong.
Thanks,
Jaeyoung
This is the control script for LAMMPS
echo both
Make the input script very simple by deleting almost all fixes/variables. Just read the data file and write dump file with ¡®run 0¡¯ to see if it gives the right geometry. Then add fixes gradually.
Sanjib
Hi, Sanjib
Thank you for your reply.
I checked my lammps scripts based on your comments.
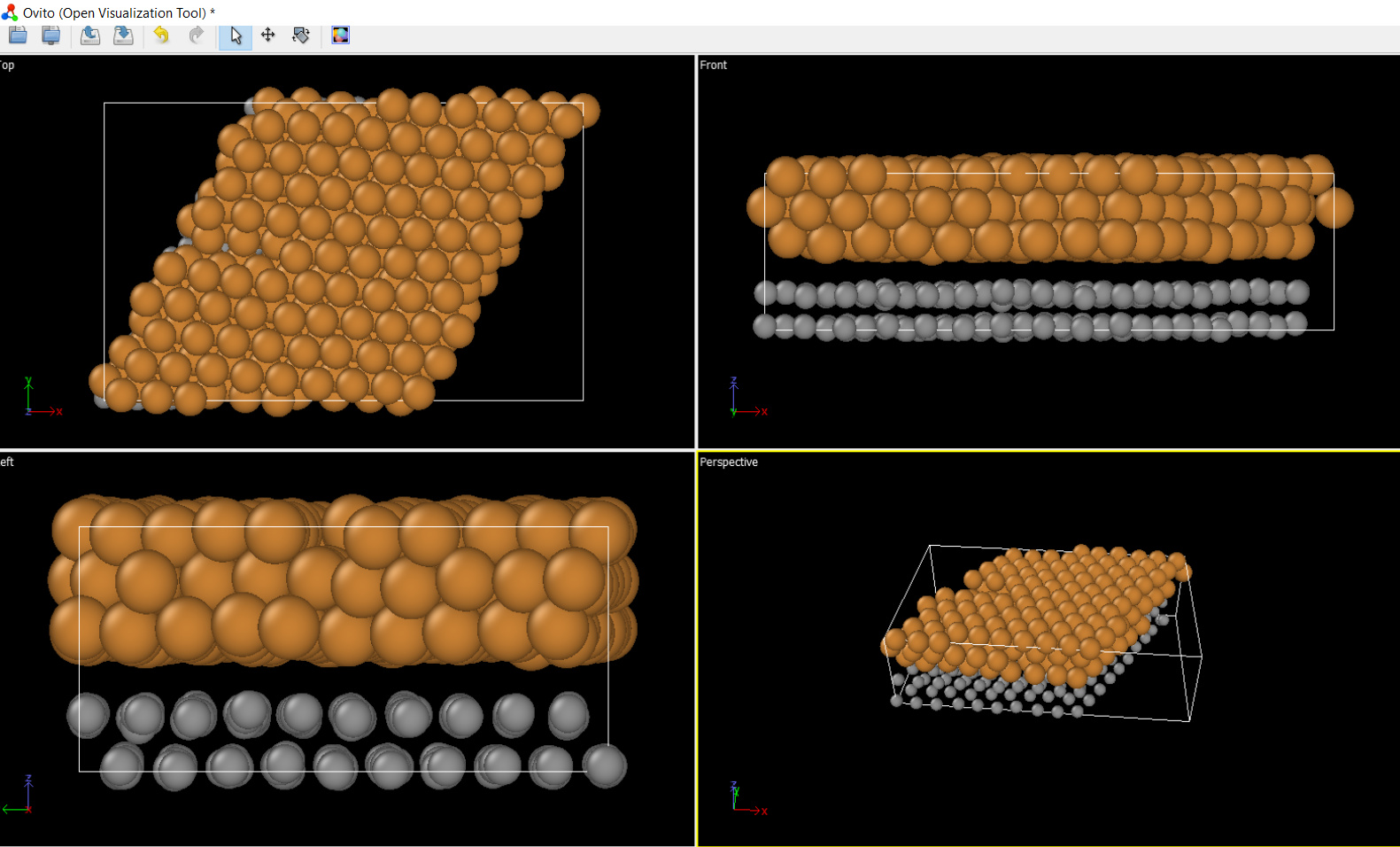
First, I ran the script with “run 0”. Its geometry was fine. Graphene layer was only single as I attached a picture.
I used this script for the first checking. I removed all fixes/variables.
However, I added the relaxation process after the first checking script and run.
Graphene geometry was changed from single layer to double layer but I don’t know where is wrong at relaxation part.
I’d appreciate it if you give me some comments.
Here is the second script with the relaxation part.
This is the control script for LAMMPS
echo both