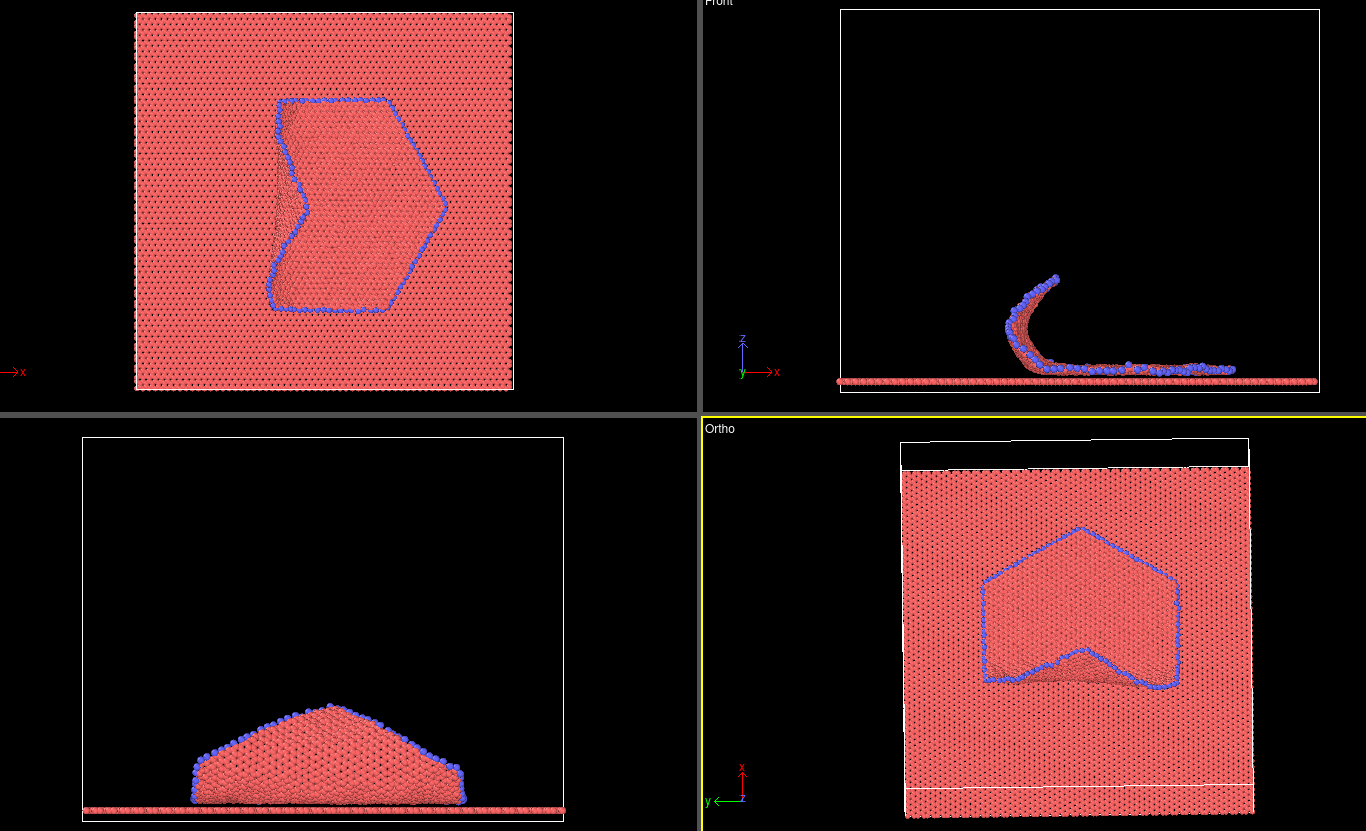
I want to study the peeling and shearing behaviour of graphene flake.
- For peeling I am giving a positive velocity of 1m/s and it is pulling the flake. For shearing I want to push flake by giving a negative velocity of 1m/s. However I am observing the same behaviour in both case. Please guide me what I can do differently. I am attaching the script that I am using.
- at the other extreme of the flake I have made the corner atoms to stay intact. However, upon visualising I can see some atoms are not in the right orientation as they tend to get detached.
initialisation
units real
dimension 3
atom_style full
bond_style harmonic
angle_style harmonic
pair_style lj/cut/coul/long 11
kspace_style pppm 1.0e-4
dihedral_style opls
improper_style harmonic
variable pre equal 1
variable tem equal 300
system definition
read_data flake1.data
read_data data.min.lammps
include PARM.lammps
simulation setting
group gcar type 1
group gbike type 2
variable zmax equal bound(gcar,zmax)-3.35
variable zmin equal bound(gcar,zmin)+3.35
region rtop block INF INF INF INF {zmax} INF
region rbot block INF INF INF INF INF {zmin}
group gtop region rtop
group gbot region rbot
variable xmax equal bound(gtop,xmax)
variable xmin equal bound(gtop,xmin)+90
region rdes block {xmin} INF INF INF {zmax} INF
group gdes region rdes
group test id 10965 11002 11003 10620 10619 10581
fix mysf2 gbot setforce 0 NULL 0
velocity gbot set 0 NULL 0
fix 6 test smd cvel 5.0 -0.00001 tether 27.664753887632 72.85058174439 6.9654 48.770636
fix tether gdes spring/self 10.0
neigh_modify every 1 delay 0 check yes
thermo_style custom step temp pe emol etotal press vol f_6[1] f_6[2] f_6[3] f_6[4] f_6[5] f_6[6] f_6[7]
dump mydmp all atom 1000 dump.lammpstrj
fix 4 all nvt temp 300.0 300.0 0.01
timestep 0.01 # (fs)
thermo 1000
run 5000000
reset_timestep 0