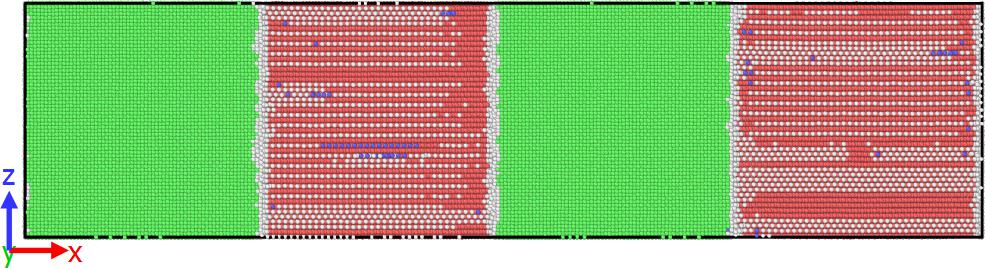
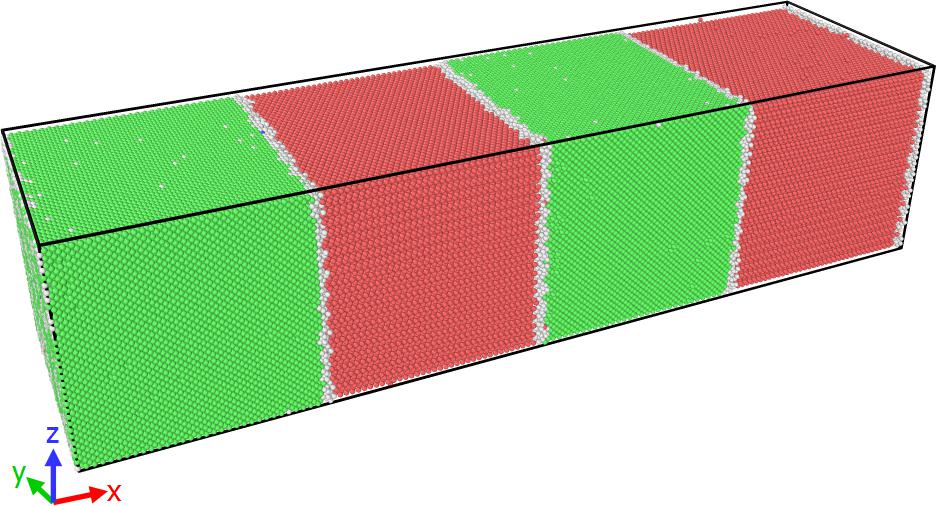
I have tried to prepare a multi-layer system of copper atoms and zirconium atoms in a cubic box.
But due to the difference in the lattice parameter of Copper (3. 61 angstroms) and Zirconium (3.23 angstroms), the specimen had defects on the Zr surface during the energy minimization process using periodic boundary conditions.
Please suggest the correct procedure to create a multi-layer specimen without surface defects.
I have attached the specimen figure showing perfect stacked Cu atoms (green) and defects (white) on the surface of Zr atoms (red).
Note: the box dimension was taken in multiples of Cu lattice parameter.
I have tried to prepare a multi-layer system of copper atoms and zirconium
atoms in a cubic box.
But due to the difference in the lattice parameter of Copper (3. 61
angstroms) and Zirconium (3.23 angstroms), the specimen had defects on the
Zr surface during the energy minimization process using periodic boundary
conditions.
Please suggest the correct procedure to create a multi-layer specimen
without surface defects.
you need to construct "commensurate" supercells.
3.61/3.23 = 1.11765 which is close to 10/9 = 1.111111
10*3.23 = 32.30
9*3.61 = 32.49
take the average of that: (32.30 + 32.49) / 2 = 32.395
this gives adjusted lattice constants of 3.60 and 3.24, respectively.
so use those lattice constants and make sure the regions are multiples 9
times the copper lattice or 10 times the zirconium, and you should have
proper slabs.
it also helps to move the region definitions to be not exactly on lattice
points, but slightly shifted, so that there are no atoms exactly on the
region boundaries and thus it would be ambiguous for LAMMPS which one to
choose. shifting by 0.1 lattice units makes those unambiguous.
I have tried to prepare a multi-layer system of copper atoms and zirconium atoms in a cubic box.
But due to the difference in the lattice parameter of Copper (3. 61 angstroms) and Zirconium (3.23 angstroms), the specimen had defects on the Zr surface during the energy minimization process using periodic boundary conditions.
Is that not what you would expect to happen?
This sounds like a physics question to me.
But it sounds like you already know the answer.
Perhaps the Zirconium atoms will retain their periodicity better if you make each Zr layer (and each Cu layer) thicker than it currently is. This should be easy to set up in LAMMPS or the tools which come with LAMMPS to build DATA files (see lammps.sandia.gov/prepost.html). I don’t know what thickness would be realistic but I doubt that single-atom-thick layers of alternating metals could exist in nature.
Thank you all for your kind response. It has been really helpful.
I have taken your suggestions and created a super cell taking 3.60 and 3.24 as lattice constants for Cu and Zr respectively. Even after that, I got some overlapping atoms.
To remove that, I used delete atom overlap command. And then I minimized it under p p p boundary conditions. For reference, I am attaching the .jpeg file of the sample (Cu-Zr multi layer).