Hi,
I am new to Materials Project and also to surface chemistry.
I am trying to reproduce the surface energy of diamond (C-mp_66) along (100) Miller Index plane using the methods discussed in VASP Surface Science tutorials. I find the POSCAR for the unit cell has different lattice parameters (along x- and y-) when compared to the (100) surface slab. I do not understand the reason.
Also, could you provide the INCAR file for slab surface energy calculation?
The Materials Project methods likely differ some from those of the VASP surface science tutorials, but if you want to generate a (100) diamond slab and VASP parameters according to the MP methods outlined in this paper, you can use the following code.
# Construct the slab
from pymatgen.core.surface import generate_all_slabs
from pymatgen import MPRester
mpr = MPRester(YOUR_API_KEY_GOES_HERE)
diamond = mpr.get_structure_by_material_id(
"mp-66", conventional_unit_cell=True)
slabs = generate_all_slabs(
structure=diamond,
max_index=1, # maximum miller index to include
min_slab_size=10.0, # slab size in angstroms
min_vacuum_size=15.0, # min vacuum size in angstroms
)
# Get the 100 slab
slab_100 = [slab for slab in slabs
if slab.miller_index == (1, 0, 0)][0]
# Construct the VASP inputs
from pymatgen.io.vasp.sets import MVLSlabSet
input_set = MVLSlabSet(slab_100)
input_set.write_input(FOLDER_TO_PUT_INPUT_IN)
# Or if you just want the incar
input_set.incar.write_file("INCAR")
Thanks for the reply. I am having an issue with generating the correct POTCAR for MP calculations. I have the original POTCAR (for VASP 5.4.4.) but that doesn’t seem to work in spite of installing pymatgen and following the procedures for POTCAR generation.
The final question is: can I scale the lattice parameters of the (100) surface .cif file (available in Materials Project for C_mp-66) according to the lattice parameters of diamond under high pressures?
I’m sorry it’s not working, I’m happy to help further if you can tell us more about what’s going wrong (the code and the error traceback is usually sufficient).
Pymatgen keeps track of a structure’s lattice in the lattice attribute. You can set this directly with a Lattice object:
# expands all lattice vectors by 1%
diamond.apply_strain(0.01)
# expands lattice vectors by 1%, 3%, and 1% respectively
diamond.apply_strain([0.01, 0.03, 0.01])
You can do this for either the bulk structure (called diamond in the code posted here) or the slab object. This is because they’re both Structure objects.
Here’s the way I try to get the POTCAR file:
Step 1. I have the python code and the VASP 5.4.4. POTCAR (original file from VASP) in Desktop
Step 2. In Anaconda Prompt:
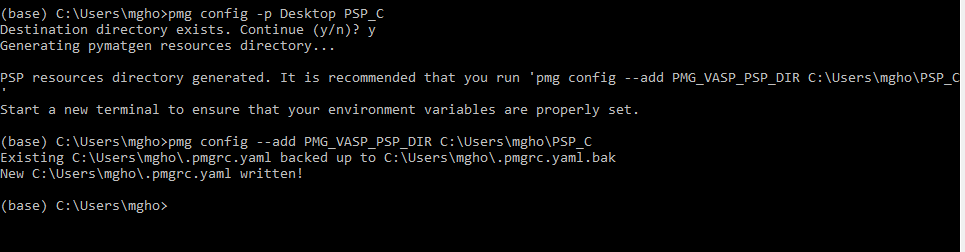
pmg config -p Desktop PSP_C
pmg config --add PMG_VASP_PSP_DIR C:\Users\mgho\PSP_C
This says that, .pmgrc.yaml is written!
Step 3. I run the python file with the code of slab generation, and the error message is: "You do not have the right POTCAR with functional PBE and label C in your VASP_PSP_DIR
I think the issue is with the first config command. Is there a sub-folder of your Desktop where the VASP-downloaded POTCARs are? If so, try issuing that instead of the “Desktop” (maybe Desktop/vasp-5.4.4/?).
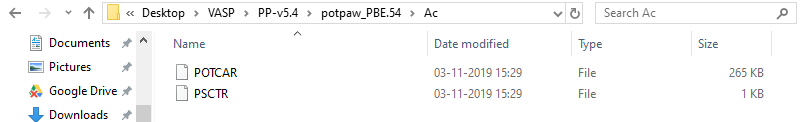
If that doesn’t fix the issue, can you post the contents of PSP_C?
No, that hadn’t fixed the issue. I initially tried making different folder which contained the python file and the VASP POSCAR file. But since it, didn’t work, I tried working in Desktop.
Pymatgen has to configure all of the POTCARs at the same time, the first argument to pmg config -p is the folder which you download from the VASP download portal and extract which contains all of the VASP POTCARs.
Oh! I initially had only one POTCAR file (the one of my interest).
I have downloaded the pseudopotential folder, PP-v54 and it contains two folders: potpaw_PBE.54 and potpaw_LDA.54. I work with PBE POTCARs and want to stick to that. So, what should be the final directory in pmg config -p command: PP-v54 or potpaw_PBE.54? I have tried the pmg config -p command for both the directories but both result into empty PSP directory (at the completion of pmg command).
You’re saying you’ve done that and that there’s nothing in the output directory, correct? I’ve replicated these steps and it’s working, but there may be an issue with windows compatibility that I’m not able to test at the moment.