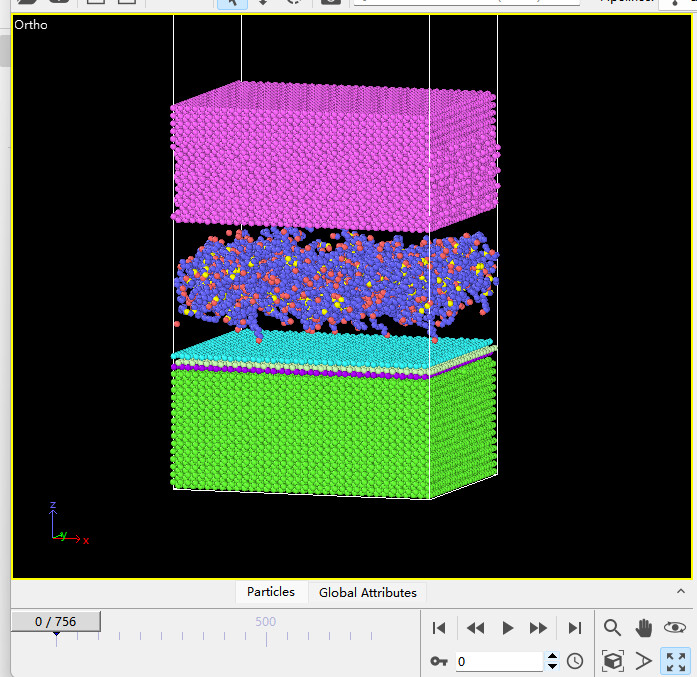
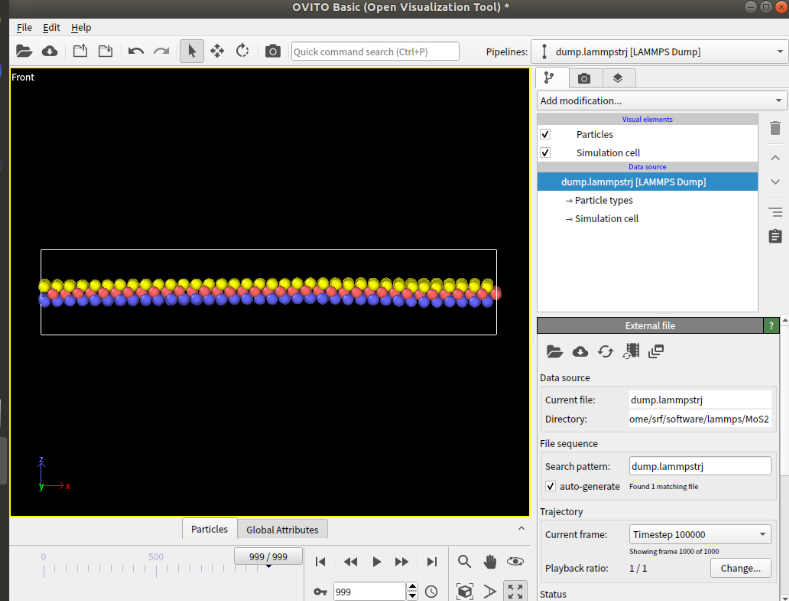
Dears all,
I am trying to create a model for Fe,PAO and MoS2.However,the MoS2’s atoms will scatter when relaxing the model.
I use the EAM potential for Fe,trappe-ua potential for PAO and sw potential for MoS2.I have tried several versions of sw potential,but all failed.
Here is my in.data.Can anyone help me correct it?
Thanks a lot.
#############################################################
units metal #energy=eV
dimension 3
boundary p p p
atom_style full
neighbor 2.0 bin
neigh_modify every 1 delay 0 check yes
bond_style harmonic
angle_style harmonic
dihedral_style opls
read_data Layer2_hybrid3.data
#define groups
group PAO type 1 2 3
group MoS2 type 6 7 8
group Fe type 4 5
region Fe_up block INF INF INF INF 100 INF
group Fe_up region Fe_up
region Fe_down block INF INF INF INF 0 43
group Fe_down region Fe_down
#define fix&thermo&free layer
region upfix block INF INF INF INF 129 INF
group upfix region upfix
region upther block INF INF INF INF 114 129
group upther region upther
region downfix block INF INF INF INF INF 15
group downfix region downfix
region downther block INF INF INF INF 15 29
group downther region downther
group upfree subtract Fe_up upfix upther
group downfree subtract Fe_down downfix downther
group fix union upfix downfix
group ther union upther downther
group free union upfree downfree
group move subtract all upfix downfix
pair_style hybrid eam/fs sw lj/cut 14
#c=type1.2.3,Fe=type4,5,Mo=type6,S=type7,8
#epsilon=sqrt(epsilon1epsilon2);sigma=0.5(sigma1+sigma2)
pair_coeff 1 1 lj/cut 0.00844 3.75000 #c3-c3
pair_coeff 1 2 lj/cut 0.00578 3.85000 #c3-c2
pair_coeff 1 3 lj/cut 0.00270 4.21500 #c3-c1
pair_coeff 1 4 lj/cut 0.01577 2.97500 #c3-Fe
pair_coeff 1 5 lj/cut 0.01577 2.97500 #c3-Fe
pair_coeff 1 6 lj/cut 0.00453 3.23450 #c3-Mo
pair_coeff 1 7 lj/cut 0.01001 3.67250 #c3-s
pair_coeff 1 8 lj/cut 0.01001 3.67250 #c3-s
pair_coeff 2 2 lj/cut 0.00396 3.95000 #c2-c2
pair_coeff 2 3 lj/cut 0.00185 4.31500 #c2-c1
pair_coeff 2 4 lj/cut 0.01080 3.07500 #c2-Fe
pair_coeff 2 5 lj/cut 0.01080 3.07500 #c2-Fe
pair_coeff 2 6 lj/cut 0.00310 3.33450 #c2-Mo
pair_coeff 2 7 lj/cut 0.00686 3.77250 #c2-s
pair_coeff 2 8 lj/cut 0.00686 3.77250 #c2-s
pair_coeff 3 3 lj/cut 0.00086 4.68000 #c1-c1
pair_coeff 3 4 lj/cut 0.00504 3.44000 #c1-Fe
pair_coeff 3 5 lj/cut 0.00504 3.44000 #c1-Fe
pair_coeff 3 6 lj/cut 0.00145 3.69950 #c1-Mo
pair_coeff 3 7 lj/cut 0.00320 4.13750 #c1-s
pair_coeff 3 8 lj/cut 0.00320 4.13750 #c1-s
pair_coeff * * eam/fs /home/srf/software/lammps/potentials/Fe_mm.eam.fs NULL NULL NULL Fe Fe NULL NULL NULL #Fe-Fe
pair_coeff 4 6 lj/cut 0.00846 2.45950 #Fe-Mo
pair_coeff 4 7 lj/cut 0.01870 2.89750 #Fe-s
pair_coeff 4 8 lj/cut 0.01870 2.89750 #Fe-s
pair_coeff 5 6 lj/cut 0.00846 2.45950 #Fe-Mo
pair_coeff 5 7 lj/cut 0.01870 2.89750 #Fe-s
pair_coeff 5 8 lj/cut 0.01870 2.89750 #Fe-s
pair_coeff * * sw /home/srf/software/lammps/potentials/tmd.sw.mod NULL NULL NULL NULL NULL Mo S S
bond_coeff 1 19.513 1.54
bond_coeff 2 19.513 1.54
bond_coeff 3 19.513 1.54
bond_coeff 4 19.513 1.54
angle_coeff 1 2.69292 114
angle_coeff 2 2.69292 114
angle_coeff 3 2.69292 114
angle_coeff 4 2.69292 114
angle_coeff 5 2.69292 114
angle_coeff 6 2.69292 114
dihedral_coeff 1 0 0.03059393 -0.00587612 0.06819027
dihedral_coeff 2 0 0.03059393 -0.00587612 0.06819027
dihedral_coeff 3 0 0.03059393 -0.00587612 0.06819027
dihedral_coeff 4 0 0.03059393 -0.00587612 0.06819027
dihedral_coeff 5 0 0.03059393 -0.00587612 0.06819027
dihedral_coeff 6 0 0.03059393 -0.00587612 0.06819027
dihedral_coeff 7 0 0.03059393 -0.00587612 0.06819027
#min
fix 1 Fe_up setforce 0 0 0
fix 2 Fe_down setforce 0 0 0
minimize 1.0e-4 1.0e-6 100 1000
unfix 1
unfix 2
#run settings
compute myTther ther temp/partial 0 1 0
compute myTfree free temp/partial 0 1 0
compute fx downfix reduce sum fx
compute fz downfix reduce sum fz
velocity ther create 300 5643454 temp myTther loop local dist gaussian rot yes
velocity free create 300 567854 temp myTfree loop local dist gaussian rot yes
timestep 0.001 #time=ps
thermo 100
thermo_style custom step etotal c_myTther c_myTfree c_fx c_fz
thermo_modify flush yes
dump mydump1 all atom 100 dump.lammpstrj
fix MoS2_nvt MoS2 nvt temp 300.0 300.0 0.1
fix PAO_nvt PAO nvt temp 300.0 300.0 0.1
fix upther_nvt upther nvt temp 300.0 300.0 0.1
fix upfree_nvt upfree nvt temp 300.0 300.0 0.1
fix downther_nvt downther nvt temp 300.0 300.0 0.1
fix downfree_nvt downfree nvt temp 300.0 300.0 0.1
restart 10000 tmp.restart
run 1000000